
МIНIСТЕРСТВО ОХОРОНИ ЗДОРОВ'Я УКРАЇНИ
НАКАЗ
м. Київ
| 04.01.2013 | N 3 |
|---|
Про внесення змiн до наказу Мiнiстерства охорони
здоров'я України вiд 26 серпня 2005 року N 426 та
визнання такими, що втратили чиннiсть, деяких
наказiв Мiнiстерства охорони здоров'я України з
питань реєстрацiї лiкарських засобiв
| Зареєстровано в Мiнiстерствi юстицiї України 15 березня 2013 р. за N 425/22957 |
Вiдповiдно до пункту 2 Порядку
державної реєстрацiї (перереєстрацiї) лiкарських
засобiв, затвердженого постановою
Кабiнету Мiнiстрiв України вiд 26 травня 2005 року
N 376, наказую:
1. Унести змiни до Порядку проведення експертизи реєстрацiйних матерiалiв на лiкарськi засоби, що подаються на державну реєстрацiю (перереєстрацiю), а також експертизи матерiалiв про внесення змiн до реєстрацiйних матерiалiв протягом дiї реєстрацiйного посвiдчення, затвердженого наказом Мiнiстерства охорони здоров'я України вiд 26 серпня 2005 року N 426, зареєстрованого в Мiнiстерствi юстицiї України 19 вересня 2005 року за N 1069/11349, виклавши його в новiй редакцiї, що додається.
2. Затвердити Положення про Квалiфiкацiйну комiсiю Мiнiстерства охорони здоров'я України, що додається.
3. Визнати такими, що втратили чиннiсть:
наказ Мiнiстерства охорони здоров'я України вiд 01 листопада 2001 року N 443 "Про реєстрацiю деяких видiв лiкарських засобiв", зареєстрований у Мiнiстерствi юстицiї України 20 листопада 2001 року за N 975/6166;
Порядок проведення державної реєстрацiї (перереєстрацiї) медичних iмунобiологiчних препаратiв в Українi, затверджений наказом Мiнiстерства охорони здоров'я України вiд 06 грудня 2001 року N 486, зареєстрований у Мiнiстерствi юстицiї України 28 лютого 2002 року за N 204/6492;
наказ Мiнiстерства охорони здоров'я України вiд 17 квiтня 2007 року N 190 "Про затвердження Порядку проведення додаткових випробувань лiкарських засобiв при проведеннi експертизи реєстрацiйних матерiалiв", зареєстрований у Мiнiстерствi юстицiї України 17 серпня 2007 року за N 949/14216;
Порядок проведення експертизи реєстрацiйних матерiалiв на лiкарськi засоби, що виробляються згiдно iз затвердженими прописами, затверджений наказом Мiнiстерства охорони здоров'я України вiд 17 грудня 2008 року N 754, зареєстрований у Мiнiстерствi юстицiї України 21 сiчня 2009 року за N 45/16061;
наказ Мiнiстерства охорони здоров'я України вiд 26 сiчня 2010 року N 55 "Про затвердження Порядку проведення експертизи матерiалiв на лiкарськi засоби обмеженого застосування, що подаються на державну реєстрацiю (перереєстрацiю)", зареєстрований у Мiнiстерствi юстицiї України 13 лютого 2010 року за N 156/17451;
наказ Мiнiстерства охорони здоров'я України вiд 17 березня 2010 року N 236 "Про затвердження Порядку проведення перевiрки виробництва лiкарських засобiв, що подаються на державну реєстрацiю", зареєстрований у Мiнiстерствi юстицiї України 13 травня 2010 року за N 323/17618.
4. Начальнику Управлiння лiкарських засобiв та медичної продукцiї (Коношевич Л. В.) забезпечити подання цього наказу на державну реєстрацiю до Мiнiстерства юстицiї України у встановленому порядку.
5. Цей наказ набирає чинностi з дня його офiцiйного опублiкування.
6. Контроль за виконанням цього наказу покласти на заступника Мiнiстра - керiвника апарату Богачева Р. М.
| В. о. Мiнiстра | О. Толстанов |
| ПОГОДЖЕНО: | |
| Голова державної служби з питань регуляторної полiтики та розвитку пiдприємництва | М. Ю. Бродський |
| Голова Державної служби України з лiкарських засобiв | О. С. Соловйов |
| ЗАТВЕРДЖЕНО Наказ Мiнiстерства охорони здоров'я України 26.08.2005 N 426 (у редакцiї наказу Мiнiстерства охорони здоров'я України вiд 04.01.2013 N 3) |
|
| Зареєстровано в Мiнiстерствi юстицiї України 15 березня 2013 р. за N 425/22957 |
ПОРЯДОК
проведення експертизи реєстрацiйних матерiалiв
на лiкарськi засоби, що подаються на державну
реєстрацiю (перереєстрацiю), а також експертизи
матерiалiв про внесення змiн до реєстрацiйних
матерiалiв протягом дiї реєстрацiйного
посвiдчення
I. ЗАГАЛЬНI ПОЛОЖЕННЯ
1.1. Цей Порядок розроблено вiдповiдно до Законiв України "Про лiкарськi засоби" та "Про захист населення вiд iнфекцiйних хвороб", Порядку державної реєстрацiї (перереєстрацiї) лiкарських засобiв i Розмiрiв збору за державну реєстрацiю (перереєстрацiю) лiкарських засобiв, затверджених постановою Кабiнету Мiнiстрiв України вiд 26 травня 2005 року N 376.
1.2. Цей Порядок проведення експертизи реєстрацiйних матерiалiв на лiкарськi засоби, що подаються на державну реєстрацiю (перереєстрацiю), а також експертизи матерiалiв про внесення змiн до реєстрацiйних матерiалiв протягом дiї реєстрацiйного посвiдчення (далi - Порядок) поширюється на активнi фармацевтичнi iнгредiєнти, продукцiю in bulk, готовi лiкарськi засоби, у тому числi медичнi iмунобiологiчнi препарати, речовини або комбiнацiї речовин, що мiстяться у засобах медичного призначення, якi у процесi використання надходять до системного кровотоку, а також засоби, якi за визначенням Мiнiстерства охорони здоров'я України (далi - МОЗ) реєструються як лiкарськi засоби (далi - лiкарськi засоби), такi як:
засоби для дезiнфекцiї шкiри та слизових оболонок носової i ротової порожнин та зовнiшнiх статевих органiв пацiєнта перед дiагностичними, оперативними манiпуляцiями чи лiкувальними процедурами;
засоби для дезiнфекцiї шкiри рук медичного персоналу до i пiсля проведення дiагностичних та оперативних манiпуляцiй i контакту з iнфiкованим та/або потенцiйно iнфiкованим матерiалом;
матерiали перев'язувальнi клейкi та iншi (тампони, марля, серветки), просоченi антисептичними або дезiнфекцiйними засобами, iншими лiкарськими засобами та упакованi для роздрiбного продажу;
засоби для знищення комах та клiщiв, якi мають епiдемiологiчне значення та призначенi для нанесення на шкiру людини та її придатки, а також репелентнi засоби для безпосереднього нанесення на шкiру людини та її придатки;
лiкарськi косметичнi засоби, якi разом з косметичною сировиною мiстять окремi дiючi речовини або їх сумiшi, що використовуються для профiлактики та/або лiкування захворювань шкiри людини та її придаткiв, слизової оболонки носової i ротової порожнин, статевих органiв пацiєнта i виробляються у формi крему, молочка, олiї, маски, бальзаму, гелю, порошку, лосьйону, шампуню, помади, екстракту для ванн, антиперспiранту, зубної пасти, елiксиру тощо;
речовини або комбiнацiї речовин, що мiстяться у засобах медичного призначення, якi у процесi використання надходять до системного кровотоку, зокрема допомiжнi засоби для гемотрансфузiй тощо.
1.3. Цей Порядок не поширюється на радiофармацевтичнi лiкарськi засоби, якi виготовленi вiдповiдно до iнструкцiй виробника, пiд час використання у акредитованих закладах охорони здоров'я, виключно iз лiцензованих джерел радiонуклiдiв, радiонуклiдних наборiв, прекурсорiв радiонуклiдiв.
1.4. Державну реєстрацiю (перереєстрацiю) лiкарського засобу здiйснює МОЗ на пiдставi результатiв експертизи реєстрацiйних матерiалiв (реєстрацiйного досьє) на такий засiб та контролю його якостi, проведених Державним експертним центром МОЗ (далi - Центр).
II. ВИЗНАЧЕННЯ ТЕРМIНIВ
У цьому Порядку наведенi нижче термiни вживаються у таких значеннях:
активний фармацевтичний iнгредiєнт (лiкарська речовина, дiюча речовина, субстанцiя) (далi - АФI або дiюча речовина) - будь-яка речовина чи сумiш речовин, що призначена для використання у виробництвi лiкарського засобу i пiд час цього використання стає його активним iнгредiєнтом. Такi речовини мають фармакологiчну чи iншу безпосередню дiю на органiзм людини. У складi готових форм лiкарських засобiв їх застосовують для лiкування, дiагностики чи профiлактики захворювання, для змiни стану, структур або фiзiологiчних функцiй органiзму, для догляду, обробки та полегшення симптомiв.
АФI або дiюча речовина можуть бути: компактнi, вкритi оболонкою, гранульованi, здрiбненi до певного ступеня або обробленi iншим шляхом та пiд рiзними назвами й формами (зокрема у формах пелет, гранул тощо та iнших формах випуску);
безпека лiкарського засобу - характеристика лiкарського засобу, яка базується на порiвняльнiй оцiнцi користi вiд його застосування та потенцiйної шкоди, яка може бути завдана пацiєнту при застосуваннi ним цього лiкарського засобу;
бiовейвер - процедура проведення порiвняльних дослiджень in vitro на пiдставi бiофармацевтичної системи класифiкацiї (далi - БСК) з використанням тесту "Розчинення" для пiдтвердження еквiвалентностi генеричного та референтного лiкарських засобiв системної дiї у твердiй дозованiй формi для орального застосування з негайним вивiльненням з метою реєстрацiї генеричного лiкарського засобу без проведення дослiджень in vivo;
бiодоступнiсть - швидкiсть i ступiнь, з якими дiюча речовина або її активний компонент абсорбується (всмоктується) з лiкарської форми i стає доступною у мiсцi дiї;
бiоеквiвалентнiсть - два лiкарських засоби бiоеквiвалентнi, якщо вони є фармацевтично еквiвалентними або фармацевтично альтернативними i якщо їхня бiодоступнiсть пiсля введення в однiй i тiй самiй молярнiй дозi подiбна до такого ступеня, що ефекти цих лiкарських засобiв щодо ефективностi та безпеки будуть по сутi однаковими;
бiологiчнi лiкарськi засоби - лiкарськi засоби, що мiстять дiючi речовини бiологiчного походження, отриманi шляхом виробництва з бiологiчного джерела (тваринного, людського, рослинного, мiкробного або бiотехнологiчного);
бiофармацевтична система класифiкацiї - наукова система класифiкацiї дiючих речовин на основi їх розчинностi у водних розчинах та ступеня кишкового проникнення;
виробник лiкарських засобiв - суб'єкт господарювання, який здiйснює хоча б один iз етапiв виробництва лiкарських засобiв та має лiцензiю (дозвiл) на виробництво лiкарських засобiв (якщо останнє передбачено нацiональним законодавством країни, на територiї якої знаходяться виробничi потужностi виробника);
виробництво лiкарських засобiв - дiяльнiсть, пов'язана iз серiйним випуском лiкарських засобiв, яка включає всi або хоча б одну зi стадiй технологiчного процесу, у тому числi закупiвлю матерiалiв i продукцiї, фасування, пакування та/або маркування, зберiгання, вiдповiдний контроль, видачу дозволу на випуск (реалiзацiю), а також оптову торгiвлю (дистрибуцiю) продукцiєю власного виробництва;
висновок Державного експертного центру МОЗ щодо ефективностi, безпеки та якостi лiкарського засобу - узагальнений результат експертизи (первинної, та/або попередньої, та/або спецiалiзованої) матерiалiв реєстрацiйного досьє на лiкарський засiб з наданням рекомендацiй щодо його державної реєстрацiї (перереєстрацiї), внесення змiн до матерiалiв реєстрацiйного досьє на лiкарський засiб протягом дiї реєстрацiйного посвiдчення чи його нової реєстрацiї або щодо вiдмови в реєстрацiї (перереєстрацiї), внесеннi змiн до матерiалiв реєстрацiйного досьє на лiкарський засiб протягом дiї реєстрацiйного посвiдчення;
високотехнологiчнi (бiотехнологiчнi) лiкарськi засоби - лiкарськi засоби, що мiстять дiючi речовини, отриманi за допомогою методiв бiотехнологiї, таких як: генно-iнженерна технологiя, клiтинна iнженерiя, гiбридомнi технологiї, iнженерна ензимологiя та iнженерна iмунологiя тощо;
вторинна упаковка - упаковка, у яку вкладається лiкарський засiб у первиннiй упаковцi i яка виконує захисну функцiю по вiдношенню до лiкарського засобу та первинної упаковки;
генеричний лiкарський засiб (генерик, взаємозамiнний) (далi - генерик) - лiкарський засiб, який має такий самий кiлькiсний та якiсний склад дiючих речовин i таку саму лiкарську форму, що й референтний препарат, та чия взаємозамiннiсть з референтним препаратом доведена вiдповiдними дослiдженнями. Рiзнi солi, простi та складнi ефiри, iзомери, сумiшi iзомерiв, комплекси або похiднi дiючої речовини вважаються однiєю i тiєю самою дiючою речовиною за умови, що вони суттєво не вiдрiзняються з точки зору безпеки та ефективностi. Рiзнi лiкарськi форми орального застосування з негайним вивiльненням дiючої речовини вважаються однiєю i тiєю самою лiкарською формою;
генно-iнженернi лiкарськi засоби - лiкарськi засоби, якi отриманi шляхом технологiй рекомбiнантних ДНК;
гомеопатичний лiкарський засiб - будь-який лiкарський засiб, виготовлений iз продуктiв, субстанцiй або складових, якi називаються гомеопатичною сировиною, вiдповiдно до процедури виготовлення гомеопатичного лiкарського засобу, описаної в Державнiй фармакопеї України (далi - ДФУ) або Європейськiй фармакопеї або - в разi вiдсутностi такого опису - у Нiмецькiй гомеопатичнiй фармакопеї (GHP), Гомеопатичнiй фармакопеї США (HPUS), Британськiй гомеопатичнiй фармакопеї (BHP), Гомеопатичнiй фармакопеї Швабе.
Гомеопатичний лiкарський засiб також може мiстити бiльше однiєї дiючої речовини;
державна перереєстрацiя лiкарського засобу - процедура, яка проводиться вiдповiдно до вимог чинного законодавства та цього Порядку з метою продовження строку, протягом якого лiкарський засiб дозволяється до медичного застосування в Українi;
державна реєстрацiя лiкарського засобу - процедура, яка проводиться вiдповiдно до вимог чинного законодавства та цього Порядку з метою допуску лiкарського засобу до медичного застосування в Українi та внесення його до Державного реєстру лiкарських засобiв України;
джерело радiонуклiдiв - будь-яка система, яка мiстить фiксований первинний радiонуклiд, з якого утворюються вториннi радiонуклiди, якi витягуються шляхом елюювання або в iнший спосiб та використовуються в радiофармацевтичному лiкарському засобi;
дiючi речовини (бiологiчнi агенти) - бiологiчно активнi речовини, у тому числi бiологiчнi агенти (мiкроорганiзми, вiруси, клiтини та тканини людини, тварин i їх компоненти), якi можуть змiнювати стан i функцiї органiзму або мають профiлактичну, дiагностичну чи лiкувальну дiю та використовуються як основний(i) компонент(и) для виробництва готових лiкарських засобiв;
добре вивчене медичне застосування - медичне застосування дiючої(их) речовини(ин), що входить(ять) до складу лiкарського засобу, добре вивчене(i), визнанi її (їх) ефективнiсть та прийнятний ступiнь безпеки (що пiдтверджено докладними бiблiографiчними посиланнями на опублiкованi данi щодо пiсляреєстрацiйних, епiдемiологiчних дослiджень тощо), i минуло не менше 10 рокiв вiд дати першого систематичного i документованого застосування в Українi дiючої(их) речовини(н) у складi лiкарського(их) засобу(iв);
додатковi вивчення (випробування) - дослiдження, необхiднiсть яких визначається пiд час проведення спецiалiзованої експертизи та якi дозволяють забезпечити складання вмотивованих висновкiв щодо ефективностi, безпеки та якостi лiкарського засобу при його реєстрацiї та/або внесеннi змiн до реєстрацiйних матерiалiв;
дослiдження еквiвалентностi - дослiдження, яке визначає еквiвалентнiсть мiж генериком та референтним лiкарським засобом при використаннi дослiджень in vivo та/або in vitro;
дослiдження еквiвалентностi in vitro на пiдставi БСК - комплекснi дослiдження, якi базуються на класифiкацiї дiючої речовини згiдно з БСК та розчиненнi лiкарського засобу, а також включають порiвняння профiлiв розчинення генерика та референтного лiкарського засобу у трьох середовищах зi значеннями pH 1,2, pH 4,5 i pH 6,8;
експерт з питань реєстрацiї Центру - фiзична особа, яка має вiдповiдний рiвень квалiфiкацiї i знань законодавства України, правил i норм Європейського Союзу, рекомендацiй ВООЗ у сферi обiгу лiкарських засобiв, положень Мiжнародної конференцiї з гармонiзацiї технiчних вимог до реєстрацiї лiкарських засобiв для людини. При цьому експерт не повинен виконувати функцiї, у тому числi за межами Центру, що призводять до конфлiкту iнтересiв;
експертиза матерiалiв реєстрацiйного досьє на лiкарський засiб - попередня та спецiалiзована експертиза матерiалiв реєстрацiйного досьє та за необхiдностi матерiалiв додаткових вивчень (випробувань) лiкарського засобу з метою пiдготовки вмотивованих висновкiв та рекомендацiй щодо прийняття рiшення про можливiсть його державної реєстрацiї (перереєстрацiї), внесення змiн до реєстрацiйних матерiалiв або щодо вiдмови в реєстрацiї (перереєстрацiї), внесеннi змiн до реєстрацiйних матерiалiв на лiкарський засiб;
ефективнiсть лiкарського засобу - сприятлива дiагностична, лiкувальна чи профiлактична дiя лiкарського засобу на встановлення характеру захворювання, його перебiг, тривалiсть або корекцiю стану чи фiзiологiчних функцiй органiзму людини вiдповiдно до показань до застосування, зазначених в iнструкцiї для медичного застосування;
загальноприйнята назва - мiжнародна непатентована назва (далi - МНН) дiючої речовини, рекомендована Всесвiтньою органiзацiєю охорони здоров'я (далi - ВООЗ), або за вiдсутностi такої звичайна загальноприйнята назва;
заява iнформованої згоди - заява про державну реєстрацiю лiкарського засобу, аналогiчного зареєстрованому, за умови, що власник реєстрацiйного посвiдчення зареєстрованого ранiше лiкарського засобу дає згоду використовувати його фармацевтичнi, доклiнiчнi та клiнiчнi данi для нової реєстрацiї лiкарського засобу з тим самим кiлькiсним та якiсним складом дiючих речовин у такiй самiй лiкарськiй формi;
заява на генерик - заява про державну реєстрацiю лiкарського засобу, який мiстить ту саму дiючу речовину i в тiй самiй лiкарськiй формi, що й референтний лiкарський засiб;
заява на лiкарський засiб з продукцiї in bulk - заява на державну реєстрацiю готового лiкарського засобу, виробництво якого здiйснюється шляхом фасування та/або кiнцевого пакування i маркування продукцiї in bulk;
заява на лiкарський засiб, який має добре вивчене медичне застосування, - заява про державну реєстрацiю лiкарського засобу, який мiстить вiдому(i) дiючу(i) речовину(и), медичне застосування якої (яких) добре вивчене в Українi протягом щонайменше 10 рокiв та яка має визнану ефективнiсть та прийнятний рiвень безпеки, що пiдтверджується докладними бiблiографiчними даними;
заява на фiксовану комбiнацiю - заява про державну реєстрацiю лiкарського засобу, що мiстить вiдомi дiючi речовини, якi не використовувались ранiше в цiй комбiнацiї, а також ранiше не використовувались у медичнiй практицi як окремi лiкарськi засоби у тiй самiй лiкарськiй формi, дозах та вiдповiдному спiввiдношеннi, що й у комбiнованому лiкарському засобi;
заявник (власник реєстрацiйного посвiдчення) (далi - заявник) - юридична або фiзична особа, яка є вiдповiдальною за ефективнiсть, якiсть та безпеку лiкарського засобу в порядку, визначеному чинним законодавством, та має ресурси для здiйснення фармаконагляду в Українi, а також є вiдповiдальною за достовiрнiсть iнформацiї, що мiститься у наданих нею матерiалах реєстрацiйного досьє;
змiни, якi можуть мати мiсце протягом дiї реєстрацiйного посвiдчення, - запропонованi заявником змiни, якi стосуються зареєстрованого (перереєстрованого) лiкарського засобу;
iнструкцiя для медичного застосування лiкарського засобу (iнструкцiя про застосування лiкарського засобу) (далi - iнструкцiя для медичного застосування) - офiцiйно затверджена iнформацiя про медичне застосування лiкарського засобу, викладена вiдповiдно до цього Порядку, що супроводжує готовий лiкарський засiб;
конфiденцiйна реєстрацiйна iнформацiя - науково-технiчна iнформацiя, що мiститься у заявi про державну реєстрацiю (перереєстрацiю) лiкарського засобу та заявi про внесення змiн до матерiалiв реєстрацiйного досьє протягом строку дiї реєстрацiйного посвiдчення та додатках до них, включаючи частини I, II, III та IV або модулi 1, 2, 3, 4 та 5 матерiалiв реєстрацiйного досьє (за винятком iнформацiї, яка є загальнодоступною, зокрема про назву лiкарського засобу, склад дiючих речовин, силу дiї, якi наводяться в межах iнструкцiї для медичного застосування, пакування, про заявника та/або виробника лiкарського засобу, iнструкцiї для медичного застосування, iнформацiї щодо небезпечних властивостей лiкарського засобу, якi можуть завдати шкоду пацiєнту пiд час застосування);
лiкарський засiб рослинного походження - будь-який лiкарський засiб, що мiстить виключно дiючу(i) речовину(и) з однiєї або бiльше рослинних субстанцiй, або один або бiльше рослинних препаратiв, або одну або бiльше рослинних субстанцiй у комбiнацiї з одним або бiльше рослинним препаратом;
лiкарський засiб, що виробляється згiдно iз затвердженим прописом, - лiкарський засiб, до якого входить дiюча речовина з добре вивченим медичним застосуванням, з визначеною ефективнiстю, задовiльним ступенем безпеки, пропис на який затверджено МОЗ;
лiкарськi засоби вiтчизняного виробництва - лiкарськi засоби, що виготовляються локальним (українським) виробником; не вважаються лiкарськими засобами вiтчизняного виробництва такi лiкарськi засоби, що пройшли тiльки стадiї фасування та/або кiнцевого пакування i маркування;
лiкарськi засоби, якi одержують з кровi або плазми людини, - лiкарськi засоби на основi компонентiв кровi; такi лiкарськi засоби включають, зокрема, альбумiн, фактори згортання кровi та iмуноглобулiни людського походження;
локальний (український) виробник - суб'єкт господарювання, який здiйснює усi технологiчнi етапи виробництва субстанцiй та/або готових лiкарських засобiв на виробничих дiльницях, розташованих на територiї України, та має лiцензiю на виробництво лiкарських засобiв;
медичнi iмунобiологiчнi препарати (далi - МIБП) - алергени, антигени, вакцини (анатоксини), цитокiни, iмуномодулятори бактерiйного походження, а також тi, що створенi на основi органiв i тканин, препарати, якi одержують з кровi та плазми людини, iмуннi сироватки, iмуноглобулiни (включаючи моноклональнi антитiла), пробiотики, iнтерферони, iншi лiкарськi засоби, призначенi для використання в медичнiй практицi з метою лiкування, специфiчної профiлактики, дiагностики стану iмунiтету (in vivo).
МIБП одержують шляхом культивування штамiв мiкроорганiзмiв i клiтин еукарiотiв, екстракцiї речовин з бiологiчних тканин, включаючи тканини людини, тварин i рослин (алергени), застосування методiв генної iнженерiї, гiбридомної технологiї, репродукцiї живих агентiв в ембрiонах чи тваринах;
назва лiкарського засобу - назва, дана лiкарському засобу, яка може бути як вигаданою заявником (виробником), так i загальноприйнятою або науковою, що може супроводжуватися назвою торгової марки або назвою заявника (виробника);
незалежний експерт - фiзична особа, яка має вiдповiдний рiвень квалiфiкацiї, спецiальнi знання i на замовлення заявника здiйснює наукову чи науково-технiчну експертизу та вiдповiдає перед заявником за достовiрнiсть i повноту аналiзу, обґрунтованiсть рекомендацiй вiдповiдно до вимог завдання заявника на проведення експертизи i не є автором, дослiдником об'єкта експертизи; яка не працює експертом у закладах або органiзацiях, що офiцiйно визначенi як експертнi органи об'єктiв наукової i науково-технiчної дiяльностi щодо обiгу лiкарських засобiв; або iншим чином пов'язана з офiцiйними експертними органами, центральним органом виконавчої влади, що забезпечує формування державної полiтики у сферi охорони здоров'я, або вповноваженими ним органами; яка визначається виключно заявником;
неiнтервенцiйне дослiдження - дослiдження, у якому лiкарськi засоби призначаються звичайним способом вiдповiдно до затвердженої iнструкцiї для медичного застосування. Залучення пацiєнта в групу з визначеним методом лiкування в протоколi клiнiчного дослiдження заздалегiдь не передбачено, а призначення лiкарського засобу диктується сучасною практикою i не залежить вiд рiшення включити пацiєнта у випробування. Не застосовують додаткових дiагностичних або монiторингових процедур щодо пацiєнтiв, а для аналiзу зiбраних даних використовують епiдемiологiчнi методи;
неправомiрне використання реєстрацiйної iнформацiї вiдносно безпеки та ефективностi лiкарського засобу - посилання або iнше використання iнформацiї про ефективнiсть та безпеку лiкарського засобу, зареєстрованого за повною i незалежною заявою, ранiше нiж за 5 рокiв з дати реєстрацiї такого лiкарського засобу в Українi, для подання заяви про державну реєстрацiю вiдповiдного генеричного лiкарського засобу, що подається для державної реєстрацiї за вiдповiдною заявою, за винятком випадкiв, коли заявник вiдповiдно до закону одержав право посилатися та/або використовувати реєстрацiйну iнформацiю референтного/оригiнального лiкарського засобу, або подав власну повну реєстрацiйну iнформацiю, що вiдповiдає вимогам до реєстрацiйної iнформацiї референтного/оригiнального лiкарського засобу.
Цi вимоги не перешкоджають суб'єкту здiйснювати в цей перiод вiдповiдну розробку лiкарського засобу, у тому числi проводити дослiдження з еквiвалентностi мiж генеричним та референтним лiкарськими засобами, для отримання реєстрацiйного посвiдчення пiсля виповнення п'яти рокiв з дати реєстрацiї в Українi референтного лiкарського засобу, визначеного в попередньому абзацi;
оригiнальний (iнновацiйний) лiкарський засiб - лiкарський засiб, що був уперше у свiтi зареєстрований на основi повної документацiї щодо його ефективностi, безпеки та якостi (повного реєстрацiйного досьє);
патентований лiкарський засiб - лiкарський засiб, який надходить в обiг пiд власною назвою виробника (заявника), право на виробництво (виготовлення), реалiзацiю та застосування якого охороняється законодавством України про охорону прав iнтелектуальної власностi;
первинна упаковка - iндивiдуальна упаковка, яка безпосередньо контактує з лiкарським засобом та сприяє збереженню його основних властивостей;
повна i незалежна заява (автономна заява) - заява на державну реєстрацiю лiкарського засобу, до якого входить нова або вiдома АФI або дiюча речовина, що зареєстрована у складi лiкарського засобу в iншiй лiкарськiй формi;
подiбний бiологiчний лiкарський засiб (бiосимiляр) - бiологiчний лiкарський засiб, подiбний щодо ефективностi, безпеки та якостi до зареєстрованого референтного бiологiчного препарату, перiод патентного захисту якого закiнчився. Подiбнiсть терапевтичної ефективностi, безпеки i якостi такого лiкарського засобу до референтного має бути доведена вiдповiдними порiвняльними доклiнiчними та клiнiчними дослiдженнями;
представник заявника (уповноважена особа, що виступає вiд iменi заявника) (далi - представник заявника) - юридична або фiзична особа, якiй на пiдставi вiдповiдного доручення заявник надав право представляти його iнтереси при проведеннi процедур реєстрацiї, перереєстрацiї та/або внесення змiн до реєстрацiйних матерiалiв на територiї України та яка є вiдповiдальною за достовiрнiсть iнформацiї, що мiститься у матерiалах реєстрацiйного досьє, наданих ним до Центру;
прекурсор радiонуклiда - будь-який радiонуклiд, призначений для введення радiоактивної мiтки до iншої речовини перед її застосуванням;
препарат обмеженого застосування (препарат-сирота) - лiкарський засiб, що призначений для дiагностики, профiлактики чи лiкування рiдкiсного захворювання, тобто захворювання, що загрожує життю чи призводить до втрати працездатностi не бiльше 5 осiб з кожних 10000 жителiв на дату подання заяви про державну реєстрацiю;
продукцiя in bulk - будь-який лiкарський засiб, що пройшов усi стадiї технологiчного процесу, за винятком стадiї фасування та/або кiнцевого пакування i маркування;
пропис (монографiя) - iнформацiя про склад, технологiю виробництва, контроль якостi та застосування лiкарського засобу;
радiонуклiдний набiр - будь-який лiкарський засiб, який має бути поєднаний або змiшаний з радiонуклiдами у готовому радiофармацевтичному лiкарському засобi, як правило, перед його застосуванням;
радiофармацевтичний лiкарський засiб - будь-який лiкарський засiб, який у готовому для застосування станi мiстить один або декiлька радiонуклiдiв (радiоактивних iзотопiв), уведених до нього з медичною метою;
регулярно оновлюваний звiт з безпеки лiкарського засобу (регулярний звiт) - письмовий звiт, який мiстить регулярно оновлювану iнформацiю з безпеки лiкарського засобу вiдповiдно до Порядку здiйснення нагляду за побiчними реакцiями лiкарських засобiв, дозволених до медичного застосування, затвердженого наказом Мiнiстерства охорони здоров'я України вiд 27 грудня 2006 року N 898, зареєстрованого у Мiнiстерствi юстицiї України 29 сiчня 2007 року за N 73/13340;
реєстрацiйна iнформацiя - науково-технiчна iнформацiя в будь-якiй формi й виглядi, збережена на будь-яких носiях, у тому числi iлюстрацiї (карти, дiаграми, органограми, малюнки, схеми тощо), фотографiї та будь-якi iншi документованi вiдомостi, що мiстяться в заявi про державну реєстрацiю (перереєстрацiю) лiкарського засобу, заявi про внесення змiн до реєстрацiйних матерiалiв протягом дiї реєстрацiйного посвiдчення та в реєстрацiйних досьє i визначена як конфiденцiйна згiдно з абзацом тридцять дев'ятим цього роздiлу;
реєстрацiйне посвiдчення на лiкарський засiб - документ, у якому мiститься iнформацiя про лiкарськiй засiб, зареєстрований в Українi та внесений до Державного реєстру лiкарських засобiв України та мiжвiдомчої бази даних про зареєстрованi в Українi лiкарськi засоби;
реєстрацiйне посвiдчення на лiкарський засiб (медичний iмунобiологiчний препарат) - документ, у якому мiститься iнформацiя про медичний iмунобiологiчний препарат, зареєстрований в Українi та внесений до Державного реєстру лiкарських засобiв України та мiжвiдомчої бази даних про зареєстрованi в Українi лiкарськi засоби;
реєстрацiйний номер - кодова позначка, яка присвоюється лiкарському засобу при державнiй реєстрацiї i зберiгається за лiкарським засобом незмiнною на весь перiод перебування лiкарського засобу на фармацевтичному ринку України;
реєстрацiйнi матерiали (матерiали реєстрацiйного досьє) - комплект документiв, що додаються до заяви про державну реєстрацiю (перереєстрацiю, внесення змiн до реєстрацiйних матерiалiв) лiкарського засобу, на пiдставi яких можна зробити обґрунтований висновок щодо його ефективностi, безпеки та якостi;
референтний лiкарський засiб - лiкарський засiб, з яким має порiвнюватися генеричний лiкарський засiб i який насамперед є оригiнальним (iнновацiйним) лiкарським засобом з доведеною ефективнiстю, безпекою та якiстю;
рослиннi препарати - препарати, одержанi у результатi обробки рослинних субстанцiй шляхом витягування, дистиляцiї, вiджимання, подрiбнення, очищення, концентрацiї та ферментацiї. Сюди входять потовченi або порошкоподiбнi рослиннi субстанцiї, настойки, екстракти, ефiрнi олiї, вiджатi соки та обробленi витяжки;
рослиннi субстанцiї - цiлi, подрiбненi або порiзанi рослини, частини рослин, водоростей, грибiв, лишайникiв у необробленiй, зазвичай засушенiй формi, iнодi свiжi. Певнi витяжки з рослин (наприклад, смоли), не призначенi для лiкування, також вважаються рослинними субстанцiями. Рослиннi субстанцiї чiтко визначаються морфологiчною частиною рослини, що використовується, та її ботанiчною назвою вiдповiдно до бiномної системи (рiд, вид, рiзновид та джерело);
сила дiї лiкарського засобу - умiст активної(их) речовини (речовин) у кiлькiсному вираженнi на одиницю дози, або одиницю об'єму, або одиницю маси вiдповiдно до лiкарської форми;
терапевтична еквiвалентнiсть - два лiкарськi засоби вважаються терапевтично еквiвалентними, якщо вони є фармацевтично еквiвалентними або фармацевтично альтернативними лiкарськими засобами i пiсля введення пацiєнтам одним i тим самим шляхом, в однаковiй молярнiй дозi їх ефекти щодо безпеки й ефективностi за суттю аналогiчнi. Це можна довести вiдповiдними дослiдженнями: порiвняльними фармакокiнетичними дослiдженнями, порiвняльними фармакодинамiчними дослiдженнями, порiвняльними клiнiчними дослiдженнями або дослiдженнями еквiвалентностi in vitro;
традицiйний лiкарський засiб - лiкарський засiб, зокрема рослинного походження, який вiдповiдає таким умовам:
лiкарський засiб вiдповiдно до його складу та призначення передбачений для застосування без нагляду лiкаря з метою дiагностики, без приписання або рецепта або без спостереження за процесом лiкування;
лiкарський засiб застосовується у певних концентрацiї та дозуваннi;
лiкарський засiб призначений для орального, зовнiшнього або iнгаляцiйного застосування;
є документальне пiдтвердження того, що лiкарський засiб застосовувався у медичнiй практицi не менше 30 рокiв у свiтi та не менше 10 рокiв в Українi;
є досить даних щодо традицiйного застосування лiкарського засобу (безпека застосування при звичайних умовах, доведена ефективнiсть);
узагальнюючий звiт - письмовий звiт, який узагальнює iнформацiю з безпеки лiкарського засобу, що мiститься у двох або бiльше регулярно оновлюваних звiтах з безпеки лiкарського засобу;
фармацевтична еквiвалентнiсть - лiкарськi препарати є фармацевтично еквiвалентними, якщо вони мають однакову лiкарську форму, вводяться тим самим шляхом, мiстять ту саму кiлькiсть тiєї самої дiючої речовини (тих самих дiючих речовин) у тих самих дозованих формах, вiдповiдають вимогам тих самих або порiвнюваних стандартiв. Фармацевтична еквiвалентнiсть передбачає терапевтичну еквiвалентнiсть, якщо лiкарська форма препарату унеможливлює вплив на бiодоступнiсть таких факторiв, як вiдмiнностi у допомiжних речовинах та/або технологiї виробництва, або iншi коливання, якi можуть впливати на швидкiсть розчинення та/або абсорбцiї дiючої речовини лiкарського засобу;
фармацевтично альтернативнi лiкарськi засоби - препарати, що мiстять однакову молярну кiлькiсть тiєї самої дiючої речовини, але вiдрiзняються за лiкарською формою (наприклад, таблетки порiвняно з капсулами) та/або хiмiчною формою (наприклад, iншi солi, iншi ефiри). Альтернативнi лiкарськi засоби доставляють ту саму дiючу речовину тим самим шляхом введення, але не є фармацевтично еквiвалентними. Вони можуть бути або можуть не бути бiоеквiвалентними або терапевтично еквiвалентними референтному препарату;
якiсть лiкарського засобу - сукупнiсть властивостей, якi надають лiкарському засобу здатнiсть задовольняти споживачiв вiдповiдно до свого призначення i вiдповiдають вимогам, встановленим законодавством.
Iншi термiни, що використовуються у цьому Порядку, вживаються у значеннях, наведених у законодавствi.
III. Порядок проведення експертизи реєстрацiйних матерiалiв на лiкарський засiб, що подається на державну реєстрацiю (перереєстрацiю)
3.1. Проведенню експертизи за бажанням заявника може передувати надання Центром безкоштовних консультацiй з питань державної реєстрацiї (перереєстрацiї) лiкарських засобiв.
3.2. Для проведення експертизи заявник подає до МОЗ за принципом "єдиного вiкна":
заяву про державну реєстрацiю лiкарського засобу, у тому числi МIБП згiдно з додатком 1 до цього Порядку (у разi надання заяви на багатокомпонентнi лiкарськi засоби в декiлькох варiацiях доз дiючих речовин, якi змiнюються непропорцiйно, заява розглядається як на рiзнi лiкарськi засоби), а у випадку традицiйних лiкарських засобiв та лiкарських засобiв, що виробляються згiдно iз затвердженими прописами, подається заява про державну реєстрацiю (перереєстрацiю) лiкарського засобу за формою, наведеною у додатку 2 до цього Порядку;
заяву про державну реєстрацiю (перереєстрацiю) АФI або дiючих речовин за формою, наведеною у додатку 3 до цього Порядку;
заяву про державну перереєстрацiю лiкарського засобу за формою, наведеною у додатку 4 до цього Порядку, а у випадку традицiйних лiкарських засобiв та лiкарських засобiв, що виробляються згiдно iз затвердженими прописами, подається заява про державну реєстрацiю (перереєстрацiю) лiкарського засобу за формою, наведеною у додатку 2 до цього Порядку.
На бажання заявника може проводитись одночасно реєстрацiя готового лiкарського засобу та дiючої речовини, що входить до складу цього лiкарського засобу, iз наданням реєстрацiйних посвiдчень про державну реєстрацiю АФI, дiючої речовини та готового лiкарського засобу за умови, що матерiали реєстрацiйного досьє подаються у форматi загального технiчного документа (далi - ЗТД), структура якого наведена у додатку 5 до цього Порядку.
Iнформацiя про подану до МОЗ заяву вноситься до електронної бази даних.
Збiр та обробка персональних даних здiйснюються вiдповiдно до вимог Закону України "Про захист персональних даних".
3.3. Проведення експертизи лiкарського засобу, що подається на державну реєстрацiю (перереєстрацiю), включає такi етапи:
первинна експертиза заяви про державну реєстрацiю лiкарського засобу з точки зору його належностi до заборонених до застосування в Українi та квалiфiкацiї типу такої заяви з метою визначення обсягу реєстрацiйних матерiалiв вiдповiдно до вимог цього Порядку;
попередня експертиза комплектностi матерiалiв реєстрацiйного досьє лiкарського засобу вiдповiдно до типу заяви та вимог, визначених у пунктi 3.5 цього роздiлу;
спецiалiзована експертиза матерiалiв реєстрацiйного досьє лiкарського засобу, що проводиться з метою складання вмотивованого висновку щодо ефективностi, безпеки та якостi лiкарського засобу. Складовою спецiалiзованої експертизи є (за потреби) додаткове вивчення (додатковi випробування).
3.4. Порядок проведення первинної експертизи.
3.4.1. За результатами розгляду заяви МОЗ протягом трьох робочих днiв надсилає до Центру лист-направлення разом iз копiєю заяви (за типом заяви, визначеним заявником).
Лист-направлення на первинну експертизу вноситься до електронної бази даних.
Пiсля отримання заяви та листа-направлення вiд МОЗ експерти з питань реєстрацiї Центру у строк до семи робочих днiв здiйснюють первинну експертизу заяви щодо можливостi реєстрацiї лiкарського засобу з точки зору його належностi до заборонених до застосування в Українi та квалiфiкацiї типу такої заяви з метою визначення обсягу реєстрацiйних матерiалiв вiдповiдно до вимог цього Порядку.
За результатами первинної експертизи заяви Центр протягом трьох робочих днiв направляє до МОЗ вiдповiднi рекомендацiї, що оформляються висновком Центру щодо первинної експертизи заяви, якi протягом 5 робочих днiв розглядаються квалiфiкацiйною комiсiєю МОЗ вiдповiдно до Положення про Квалiфiкацiйну комiсiю Мiнiстерства охорони здоров'я України, затвердженого наказом Мiнiстерства охорони здоров'я України вiд 04 сiчня 2013 року N 3. Висновок щодо первинної експертизи заяви вноситься до електронної бази даних.
3.4.2. За результатами розгляду квалiфiкацiйною комiсiєю МОЗ висновку Центру щодо первинної експертизи заяви МОЗ протягом трьох робочих днiв надає заявнику письмову вiдповiдь та у разi встановлення можливостi реєстрацiї (перереєстрацiї) заявленого лiкарського засобу оформляє лист-направлення на попередню та спецiалiзовану експертизу до Центру i рекомендує заявнику надати матерiали реєстрацiйного досьє до Центру для здiйснення їх експертизи. Лист-направлення МОЗ на попередню та спецiалiзовану експертизу вноситься до електронної бази даних.
На пiдставi укладеного мiж Центром i заявником договору проводиться експертиза наданих матерiалiв реєстрацiйного досьє.
Якщо МОЗ за результатами квалiфiкацiї заяви встановив, що заявник має надати iншу заяву, йому рекомендується разом з матерiалами реєстрацiйного досьє надати нову заяву, визначену у пунктi 3.2 цього роздiлу i наведену у вiдповiдних додатках до цього Порядку, яка направляється до Центру для початку попередньої експертизи вiдповiдно до пункту 3.5 цього роздiлу.
У разi незгоди заявника щодо рекомендованого типу заяви заявник має право звернутися до МОЗ разом iз додатковими матерiалами або оскаржити рiшення МОЗ у встановленому законодавством порядку.
3.4.3. Первинна експертиза заяви на лiкарський засiб, поданий з метою його державної перереєстрацiї, не здiйснюється.
3.5. Порядок проведення попередньої експертизи.
3.5.1. Пiсля отримання листа-направлення МОЗ на проведення попередньої та спецiалiзованої експертизи матерiалiв реєстрацiйного досьє лiкарського засобу заявником пiсля оплати державного збору за реєстрацiю (перереєстрацiю) лiкарського засобу та вартостi експертних робiт подаються до Центру матерiали реєстрацiйного досьє з урахуванням рекомендацiй квалiфiкацiйної комiсiї МОЗ щодо типу заяви та вимог:
роздiлiв IX та X цього Порядку та додатка 5 до цього Порядку - для реєстрацiї;
роздiлу XII цього Порядку - для гомеопатичних лiкарських засобiв;
роздiлiв XIII та XIV цього Порядку - для традицiйних лiкарських засобiв або лiкарських засобiв, якi виробляються згiдно iз затвердженим прописом;
роздiлiв IX, X, XI цього Порядку та додатка 6 до цього Порядку - у разi реєстрацiї АФI або дiючої речовини.
Реєстрацiйнi матерiали подаються вiдповiдно до вимог роздiлу VI цього Порядку.
У разi перереєстрацiї надається комплект матерiалiв реєстрацiйного досьє вiдповiдно до додатка 7 до цього Порядку. Для традицiйних лiкарських засобiв або лiкарських засобiв, якi виробляються згiдно iз затвердженим прописом, надається комплект матерiалiв реєстрацiйного досьє вiдповiдно до додатка 8 до цього Порядку.
Реєстрацiйнi матерiали для перереєстрацiї можуть подаватися вiдповiдно до додатка 5 або 9 до цього Порядку з урахуванням вимог роздiлiв VII, VIII, IX та X цього Порядку (залежно вiд обраного заявником формату матерiалiв реєстрацiйного досьє).
У разi державної реєстрацiї (перереєстрацiї) лiкарських засобiв, якi належать до групи лiкарських засобiв "Медичнi гази", до МОЗ подається заява згiдно з додатком 2 до цього Порядку, матерiали реєстрацiйного досьє подаються вiдповiдно до перелiку, наведеного у додатку 6 до цього Порядку, доповненого iнструкцiєю для медичного застосування, з урахуванням вимог роздiлу XVI цього Порядку.
Представник заявника має гарантувати достовiрнiсть iнформацiї, наведеної у наданих матерiалах реєстрацiйного досьє.
Якщо пiд час експертизи матерiалiв реєстрацiйного досьє, поданих на державну перереєстрацiю, виникає потреба внесення додаткових змiн, заявник надає гарантiйний лист щодо сплати вартостi експертизи таких змiн.
За наявностi зауважень Центру щодо внесення змiн до матерiалiв реєстрацiйного досьє генерика, поданого на реєстрацiю (перереєстрацiю), зокрема iнструкцiї для медичного застосування, з метою приведення її у вiдповiднiсть до затвердженої iнструкцiї для медичного застосування оригiнального (референтного) лiкарського засобу, допускається вiдстрочення оплати змiн, що запропонованi Центром, без зупинки експертних робiт.
При цьому заявник має або надати обґрунтування вiдсутностi необхiдностi внесення запропонованих Центром змiн, або сплатити вартiсть проведення експертних робiт за запропонованi змiни протягом 30 календарних днiв з дати надання Центром зауважень щодо необхiдностi внесення змiн до iнструкцiї для медичного застосування генеричного лiкарського засобу.
Якщо Центром ранiше здiйснювалася експертиза доклiнiчних та/або клiнiчних матерiалiв, заявник оплачує тiльки експертнi роботи щодо додаткових матерiалiв, якщо такi є.
У разi коли заявник протягом 3 мiсяцiв з дати офiцiйного отримання рекомендацiї квалiфiкацiйної комiсiї МОЗ не подає до Центру вiдповiдних матерiалiв реєстрацiйного досьє, Центр надає МОЗ рекомендацiї щодо зняття заяви з розгляду. МОЗ протягом 5 робочих днiв розглядає наданi рекомендацiї та приймає рiшення щодо вiдмови у державнiй реєстрацiї, що затверджується наказом МОЗ. Про прийняте рiшення МОЗ у строк до 10 робочих днiв письмово повiдомляє заявника. При цьому реєстрацiйний збiр та вартiсть проведення експертних робiт заявниковi не повертаються, але за бажанням заявника можуть бути використанi для оплати iнших його експертних робiт у Центрi.
Надалi така заява може бути подана до МОЗ в установленому порядку.
3.5.2. Попередня експертиза матерiалiв реєстрацiйного досьє на лiкарський засiб, що подається на державну реєстрацiю (перереєстрацiю), починається з дати отримання матерiалiв реєстрацiйного досьє, сформованих вiдповiдно до обраного заявником та рекомендованого МОЗ типу заяви. Iнформацiя про це вноситься до електронної бази даних.
Попередню експертизу матерiалiв реєстрацiйного досьє лiкарського засобу здiйснюють експерти з питань реєстрацiї Центру.
Термiн вiдстрочення надання засвiдченої копiї документа, що пiдтверджує вiдповiднiсть умов виробництва лiкарського засобу вимогам до виробництва лiкарських засобiв в Українi (належної виробничої практики), не входить до встановлених законодавством строкiв проведення експертизи реєстрацiйних матерiалiв. Попередня експертиза реєстрацiйних матерiалiв здiйснюється протягом 14 робочих днiв. За результатами попередньої експертизи Центр надає заявнику письмову вiдповiдь та заносить вiдповiдну iнформацiю до електронної бази даних.
3.5.3. При негативних результатах попередньої експертизи Центр письмово повiдомляє заявника про те, що матерiали реєстрацiйного досьє не можуть бути направленi на спецiалiзовану експертизу, докладно зазначивши пiдстави з посиланням на номери роздiлiв, глав, статей, пунктiв цього Порядку, або одноразово запитує у заявника додатковi данi та/або iнформацiю, необхiднi для забезпечення вiдповiдностi матерiалiв реєстрацiйного досьє вимогам, встановленим цим Порядком. Копiя такого листа направляється до МОЗ та вноситься до електронної бази даних.
Заявник має надати додатковi данi та/або iнформацiю згiдно iз зауваженнями Центру у строк до 90 робочих днiв у разi реєстрацiї або в строк до 60 робочих днiв у разi перереєстрацiї або листа з обґрунтуванням термiнiв, необхiдних для їх доопрацювання. Ця iнформацiя вноситься протягом 3 робочих днiв до електронної бази даних.
Час, потрiбний для пiдготовки та подання додаткових даних та/або iнформацiї, не входить до строку проведення попередньої експертизи.
Центр має прийняти доопрацьованi заявником матерiали реєстрацiйного досьє протягом 3 робочих днiв пiсля звернення заявника та внести отриману iнформацiю до електронної бази даних.
Якщо заявник не надає Центру додатковi данi та/або iнформацiю або надає їх у неповному обсязi, а також якщо наданi заявником додатковi данi та/або iнформацiя не забезпечують вiдповiдностi матерiалiв реєстрацiйного досьє встановленим вимогам (за винятком засвiдченої копiї документа, що пiдтверджує вiдповiднiсть умов виробництва лiкарського засобу вимогам до виробництва лiкарських засобiв в Українi (належної виробничої практики)), то Центр надає рекомендацiї МОЗ щодо вiдмови в державнiй реєстрацiї (перереєстрацiї) лiкарського засобу.
МОЗ протягом 5 робочих днiв розглядає таку рекомендацiю на квалiфiкацiйнiй комiсiї i про прийняте рiшення повiдомляє заявника в письмовiй формi у строк до 10 робочих днiв. Рiшення МОЗ про вiдмову в державнiй реєстрацiї (перереєстрацiї) лiкарського засобу може бути оскаржене у встановленому законодавством порядку.
При цьому реєстрацiйний збiр та вартiсть проведених експертних робiт заявниковi не повертаються. Надалi на бажання заявника матерiали з урахуванням зауважень Центру подаються на державну реєстрацiю (перереєстрацiю) лiкарського засобу в установленому порядку.
3.5.4. У разi вiдповiдностi матерiалiв реєстрацiйного досьє лiкарського засобу вiдповiдно до типу заяви та вимог роздiлiв Порядку та додаткiв до цього Порядку, визначених у пiдпунктi 3.5.1 цього пункту, встановлених за результатами попередньої експертизи матерiалiв реєстрацiйного досьє, вони направляються на спецiалiзовану експертизу, про що Центр письмово повiдомляє заявника та вносить вiдповiдну iнформацiю до електронної бази даних.
3.6. Порядок проведення спецiалiзованої експертизи.
3.6.1. Спецiалiзовану експертизу матерiалiв реєстрацiйного досьє лiкарського засобу здiйснюють експерти з питань реєстрацiї Центру iз залученням позаштатних експертiв та консультантiв, метою якої є складання вмотивованого висновку щодо його ефективностi, безпеки та якостi, у тому числi експертиза методiв аналiзу, якi мiстяться у проектi методiв контролю якостi (далi - МКЯ), щодо дотримання вимог ДФУ та/або Європейської фармакопеї, а за вiдсутностi вимог у цих фармакопеях - iнших провiдних фармакопей (Британська фармакопея, фармакопея США, фармакопея Японiї).
Якщо заявник керується вимогами нацiональної фармакопеї або iнших фармакопей, стандарти яких вiдповiдають стандартам ДФУ, Європейської фармакопеї, то посилання на такi фармакопеї є прийнятними та не потребують внесення змiн до матерiалiв реєстрацiйного досьє з метою приведення у вiдповiднiсть до ДФУ або Європейської фармакопеї. Ця iнформацiя має бути вiдображена експертом з питань реєстрацiї Центру, що надає рекомендацiї МОЗ у частинi якостi лiкарського засобу, у своєму експертному висновку.
Якщо лiкарський засiб, на який подано заяву про державну реєстрацiю (перереєстрацiю), був зареєстрований регуляторними органами держав, якi керуються високими стандартами до якостi, що вiдповiдають стандартам, рекомендованим ВООЗ, та наданi матерiали реєстрацiйного досьє були оцiненi у рамках наведеної процедури такими регуляторними органами, вiд заявника не вимагається вносити змiни до МКЯ з посиланням на стандарти, що застосовуються в Українi. При цьому будь-якi вiдмiнностi у специфiкацiї лiкарського засобу з фармакопейними вимогами заявник має обґрунтувати.
3.6.2. Пiд час проведення спецiалiзованої експертизи матерiалiв реєстрацiйного досьє з метою одержання додаткових даних щодо ефективностi, безпеки та якостi лiкарського засобу Центр може дворазово запитати у заявника необхiднi додатковi матерiали з посиланням на номери роздiлiв, глав, статей, пунктiв цього Порядку, при цьому не допускається нових запитiв щодо матерiалiв, якi вже розглядалися експертом, у тому числi пiд час складання висновкiв за результатами експертизи. Зауваження до матерiалiв реєстрацiйного досьє, що викладаються у запитах Центру до заявника, вносяться до електронної бази даних.
Центр має прийняти доопрацьованi матерiали протягом 3 робочих днiв пiсля звернення заявника та внести отриману iнформацiю до електронної бази.
Якщо заявник протягом 90 робочих днiв пiсля другого запиту Центру щодо додаткових матерiалiв не надсилає цих матерiалiв, або не обґрунтовує iнший строк надання матерiалiв, або надає їх у неповному обсязi (за винятком засвiдченої копiї документа, що пiдтверджує вiдповiднiсть умов виробництва лiкарського засобу вимогам до виробництва лiкарських засобiв в Українi (належної виробничої практики)), Центр надає рекомендацiї МОЗ щодо вiдмови в державнiй реєстрацiї (перереєстрацiї) лiкарського засобу.
МОЗ протягом 5 робочих днiв розглядає таку рекомендацiю на квалiфiкацiйнiй комiсiї i про прийняте МОЗ рiшення, що затверджується наказом, повiдомляє заявника в письмовiй формi у строк до 10 робочих днiв. Рiшення про вiдмову в державнiй реєстрацiї (перереєстрацiї) лiкарського засобу може бути оскаржене у встановленому законодавством порядку.
При цьому реєстрацiйний збiр та вартiсть проведення експертних робiт заявниковi не повертаються. Надалi, на бажання заявника, матерiали реєстрацiйного досьє з урахуванням зауважень Центру подаються на державну реєстрацiю (перереєстрацiю) лiкарського засобу в установленому порядку.
3.6.3. Пiд час проведення спецiалiзованої експертизи матерiалiв реєстрацiйного досьє Центр може рекомендувати МОЗ направити лiкарський засiб на додатковi випробування та/або додаткову експертизу згiдно з роздiлами XIX та XX цього Порядку. Квалiфiкацiйна комiсiя МОЗ протягом 5 робочих днiв розглядає наданi матерiали та надає вiдповiднi рекомендацiї МОЗ. Про прийняте рiшення МОЗ у строк 10 робочих днiв письмово повiдомляє заявника.
Додатковi випробування можуть включати випробування щодо: фармацевтичної розробки лiкарського засобу, доклiнiчних дослiджень, ефективностi та/або безпеки лiкарського засобу, бiодоступностi та еквiвалентностi генеричних лiкарських засобiв.
Додатковi випробування здiйснюються пiсля оплати їх вартостi, установленої договором мiж заявником та закладом охорони здоров'я/установою, що проводить такi випробування.
Результати додаткових випробувань подаються до Центру для проведення їх спецiалiзованої експертизи.
Час, потрiбний для проведення додаткових випробувань, та їх вартiсть не входять до строку i вартостi експертних робiт.
Про завершення спецiалiзованої експертизи Центр iнформує заявника письмово.
3.7. Пiсля завершення проведення спецiалiзованої експертизи Центр протягом 5 робочих днiв складає та направляє до МОЗ умотивованi висновки щодо ефективностi, безпеки та якостi лiкарського засобу, до яких додає засвiдчену копiю документа, що пiдтверджує вiдповiднiсть умов виробництва лiкарського засобу, поданого на реєстрацiю (перереєстрацiю), вимогам до виробництва лiкарських засобiв в Українi, виданого Державною службою України з лiкарських засобiв, i рекомендує або не рекомендує лiкарський засiб до державної реєстрацiї (перереєстрацiї).
Iнформацiя про складання та направлення до МОЗ умотивованих висновкiв щодо ефективностi, безпеки та якостi лiкарського засобу, а також самi висновки вносяться до електронної бази даних.
Центр також рекомендує МОЗ до затвердження iнструкцiю для медичного застосування на лiкарський засiб за формою, наведеною у додатку 10 до цього Порядку, фармакопейну статтю або методи контролю якостi, що мiстять текст маркування упаковки, та рекомендує до погодження технологiчний регламент або технологiю виробництва. За бажанням заявника Центр може рекомендувати МОЗ до затвердження коротку характеристику на лiкарський засiб.
На пiдставi поданих Центром висновкiв та рекомендацiй МОЗ у мiсячний строк пiсля їх отримання приймає рiшення, що затверджується наказом МОЗ про державну реєстрацiю (перереєстрацiю) лiкарського засобу або про вiдмову в державнiй реєстрацiї (перереєстрацiї) лiкарського засобу, i розмiщує цей наказ на своєму офiцiйному сайтi.
Рiшенням про державну реєстрацiю затверджуються фармакопейна стаття або методи контролю якостi лiкарського засобу, що мiстять текст маркування первинної та вторинної (за наявностi) упаковок, iнструкцiя для медичного застосування лiкарського засобу, здiйснюється погодження технологiчного регламенту або технологiї виробництва лiкарського засобу, а також лiкарському засобу присвоюється реєстрацiйний номер, який вноситься до Державного реєстру лiкарських засобiв та мiжвiдомчої бази даних про зареєстрованi в Українi лiкарськi засоби. До Державного реєстру заноситься iнформацiя щодо можливостi рекламування лiкарського засобу.
Зберiгання документiв, наведених у цьому Порядку та додатках до нього, якi надаються заявником з метою державної реєстрацiї (перереєстрацiї), здiйснюється Центром.
Видача затверджених МОЗ при реєстрацiї (перереєстрацiї) лiкарського засобу документiв на лiкарський засiб здiйснюється МОЗ за принципом "єдиного вiкна".
3.8. Лiкарський засiб не може бути рекомендований до державної реєстрацiї, якщо за результатами спецiалiзованої експертизи не пiдтвердились висновки щодо його ефективностi, безпеки та якостi, а саме:
лiкарський засiб шкiдливий для здоров'я людини (переважання ризику вiд застосування лiкарського засобу над очiкуваною користю) та/або терапевтична ефективнiсть засобу вiдсутня за умов застосування згiдно з iнструкцiєю для медичного застосування;
склад лiкарського засобу не вiдповiдає зазначеному в матерiалах реєстрацiйного досьє;
матерiали реєстрацiйного досьє, що додаються до заяви, не вiдповiдають вимогам цього Порядку та додаткiв до нього;
якщо пiд час проведення спецiалiзованої експертизи матерiалiв реєстрацiйного досьє на лiкарський засiб, що мiстить ту саму дiючу речовину, що й референтний/оригiнальний лiкарський засiб, було виявлено, що використано або посилались на iнформацiю щодо ефективностi та безпеки цього референтного/оригiнального лiкарського засобу, зареєстрованого в Українi вперше на пiдставi поданої в повному обсязi (повної) реєстрацiйної iнформацiї, у перiод ранiше нiж за 5 рокiв з дня першої реєстрацiї референтного/оригiнального лiкарського засобу в Українi. Зазначена вимога не поширюється на випадки, коли заявник вiдповiдно до закону одержав право посилатися та/або використовувати реєстрацiйну iнформацiю референтного/оригiнального лiкарського засобу або подав власну повну реєстрацiйну iнформацiю, що вiдповiдає вимогам до реєстрацiйної iнформацiї референтного/оригiнального лiкарського засобу, або в iнших випадках, передбачених статтею 9 Закону України "Про лiкарськi засоби";
якщо внаслiдок такої реєстрацiї будуть порушенi захищенi патентом чиннi майновi права iнтелектуальної власностi, у тому числi при виробництвi, використаннi, продажу лiкарських засобiв. Вiдомостi про факт такого порушення надаються МОЗ та Центру.
3.9. Центром у разi наявностi пiдстав щодо неможливостi державної перереєстрацiї лiкарського засобу подаються вiдповiднi висновки та рекомендацiї до квалiфiкацiйної комiсiї МОЗ. За вiдсутностi таких пiдстав Центр укладає договiр iз заявником щодо експертизи матерiалiв реєстрацiйного досьє про перереєстрацiю та проводить попередню та спецiалiзовану експертизи вiдповiдно до вимог цього Порядку.
3.10. Лiкарський засiб не може бути рекомендований до державної перереєстрацiї, якщо пiд час проведення експертизи матерiалiв реєстрацiйного досьє за результатами фармаконагляду було встановлено, що лiкарський засiб шкiдливий для здоров'я людини (переважання ризику вiд застосування лiкарського засобу над очiкуваною користю) та/або терапевтична ефективнiсть засобу вiдсутня за умови застосування згiдно з iнструкцiєю для медичного застосування.
У перереєстрацiї також може бути вiдмовлено, якщо матерiали реєстрацiйного досьє не вiдповiдають вимогам, встановленим цим Порядком.
Про прийняте рiшення щодо вiдмови у перереєстрацiї МОЗ у строк до 10 робочих днiв надсилає заявнику письмову мотивовану вiдповiдь.
3.11. За наявностi об'єктивних та обґрунтованих причин i у випадках, викладених у пунктi 4.7 глави 4 роздiлу VII цього Порядку та у главi 5 роздiлу X цього Порядку, пiсля консультацiй iз заявником лiкарський засiб може бути рекомендований до державної реєстрацiї за умови додержання заявником таких зобов'язань:
продовжувати дослiдження щодо лiкарського засобу пiсля реєстрацiї;
повiдомляти про побiчнi реакцiї на лiкарський засiб.
3.12. У разi незгоди з результатами попередньої та/або спецiалiзованої експертизи заявник може оскаржити рiшення Центру до МОЗ протягом 30 робочих днiв з дати його одержання. Вiдповiднi матерiали для обґрунтування оскарження мають бути наданi заявником до МОЗ протягом 30 робочих днiв пiсля подання такого оскарження.
Центр за направленням МОЗ здiйснює спецiалiзовану експертизу наданих заявником додаткових матерiалiв у строк до 60 робочих днiв з дати їх одержання та надає вiдповiднi обґрунтованi рекомендацiї МОЗ. МОЗ у мiсячний строк приймає вiдповiдне рiшення, про що iнформує письмово заявника.
3.13. За бажанням заявника Центр надає заявнику доступ до окремих роздiлiв електронної бази даних у частинi лiкарських засобiв, поданих за направленням МОЗ на експертизу до Центру. Умови доступу заявника до окремих роздiлiв електронної бази визначаються умовами договору на проведення експертизи, укладеного мiж заявником та Центром.
IV. ПОРЯДОК ПРОВЕДЕННЯ ЕКСПЕРТИЗИ МАТЕРIАЛIВ ПРО ВНЕСЕННЯ ЗМIН ДО МАТЕРIАЛIВ РЕЄСТРАЦIЙНОГО ДОСЬЄ НА ЛIКАРСЬКИЙ ЗАСIБ
4.1. Проведенню експертизи за бажанням заявника передує надання Центром безкоштовних консультацiй з питань внесення змiн до реєстрацiйних матерiалiв на лiкарський засiб.
4.2. Заявник зобов'язаний повiдомити МОЗ про будь-якi змiни, наведенi у додатку 11 до цього Порядку та в пiдпунктах 4.2.2 - 4.2.4, 4.4.4 цього роздiлу, щодо зареєстрованого лiкарського засобу з наданням вичерпної iнформацiї про причини цих змiн та їх можливий вплив на ефективнiсть, безпеку, якiсть лiкарського засобу i внести вiдповiднi змiни до матерiалiв реєстрацiйного досьє.
4.2.1. За характером змiни класифiкують на:
змiни типу IA - незначнi змiни, що виявляють незначний вплив або не виявляють впливу на якiсть, безпеку та ефективнiсть лiкарського засобу, якi стосуються внесення поправок до змiсту матерiалiв реєстрацiйного досьє, поданих на момент прийняття рiшення про реєстрацiю (перереєстрацiю) лiкарського засобу, i не потребують його нової реєстрацiї;
змiни типу IБ - незначнi змiни, якi не можуть бути змiнами типу IA та типу II i не потребують нової реєстрацiї;
змiни типу II - будь-якi змiни до матерiалiв реєстрацiйного досьє, що не потребують нової реєстрацiї лiкарського засобу та можуть виявляти значний вплив на його якiсть, безпеку та ефективнiсть, але не можуть розглядатися як змiни типу IA та IБ.
4.2.2. Термiновi змiни, що стосуються безпеки лiкарського засобу, - термiновi тимчасовi обмеження, пов'язанi з безпекою використання лiкарського засобу, якi впроваджуються заявником у разi виявлення ризику для здоров'я людини при застосуваннi зареєстрованого (перереєстрованого) лiкарського засобу. Про кожну причину, характер i дату передбачуваного введення в дiю тимчасових термiнових обмежень заявник повiдомляє негайно Центр. У разi якщо протягом 24 годин з моменту отримання такої iнформацiї в Центру не виникає заперечень, заявник вводить у дiю цi термiновi обмеження i одночасно не пiзнiше нiж через 48 годин вiд моменту прийняття рiшення щодо введення термiнових змiн подає заяву на внесення таких змiн згiдно з цим Порядком.
У разi якщо заявнику стало вiдомо про виявленi небезпечнi властивостi лiкарського засобу для здоров'я або життя людини, що стали пiдставою для прийняття заявником рiшення про введення обмежень до його застосування, заявник повiдомляє Центр у будь-який спосiб та подає заяву про змiни, пов'язанi з виявленими проблемами з безпеки, разом з вiдповiдною документацiєю щодо цих виявлених проблем з безпеки негайно, але не пiзнiше нiж протягом 15 календарних днiв вiд дати отримання цiєї iнформацiї. Запропонованi змiни затверджуються вiдповiдним наказом МОЗ, пiсля чого лiкарський засiб має застосовуватись вiдповiдно до змiненої iнструкцiї для медичного застосування.
У разi якщо Центр отримав обґрунтовану iнформацiю про виявленi небезпечнi властивостi лiкарського засобу для здоров'я або життя людини, що стали пiдставою для прийняття вiдповiдного рiшення про введення обмежень для застосування лiкарського засобу в установленому законодавством порядку, Центр повiдомляє про це заявника у будь-який спосiб негайно, але не пiзнiше нiж протягом 15 календарних днiв з дати прийняття такого рiшення.
У разi згоди заявника з прийнятим рiшенням вiн повинен негайно, але не пiзнiше нiж протягом 15 календарних днiв з дати отримання цiєї iнформацiї подати до МОЗ заяву про внесення змiн щодо безпеки лiкарського засобу до реєстрацiйних матерiалiв. Запропонованi змiни затверджуються вiдповiдним наказом МОЗ, пiсля чого лiкарський засiб повинен вироблятися та застосовуватись вiдповiдно до змiненої iнструкцiї для медичного застосування.
У разi незгоди заявника з прийнятим рiшенням та його вiдмови вiд унесення змiн щодо безпеки лiкарського засобу до матерiалiв реєстрацiйного досьє без наданих обґрунтувань або вiдсутностi його реагування стосовно цього рiшення МОЗ приймає рiшення про заборону (тимчасову заборону) застосування лiкарського засобу шляхом припинення дiї реєстрацiйного посвiдчення у порядку, передбаченому чинним законодавством.
У разi якщо 15-й календарний день припадає на вихiдний або святковий день, iнформацiя повинна бути надана в перший пiсля нього робочий день.
4.2.3. Технiчна помилка - невiдповiдностi, допущенi у затверджених при реєстрацiї (перереєстрацiї) документах. Це не потребує проведення експертизи експертами з питань реєстрацiї Центру та виявляється при порiвняннi затверджених при реєстрацiї (перереєстрацiї) документiв з матерiалами реєстрацiйного досьє заявника. До технiчних помилок належать:
а) помилки, пов'язанi з орфографiчними та/або граматичними помилками, в тому числi з транслiтерацiєю;
б) помилки, пов'язанi з некоректним перекладом на українську та/або iншу мову адмiнiстративних даних, таких як форма власностi, найменування, мiсцезнаходження заявника/виробника лiкарського засобу;
в) невiдповiднiсть у МКЯ, що мiстить маркування первинної та вторинної (за наявностi) упаковок лiкарського засобу, короткiй характеристицi лiкарського засобу, iнструкцiї для медичного застосування лiкарського засобу;
г) помилки у МКЯ, пов'язанi з перекладом або перенесенням iнформацiї щодо:
назви, кiлькiсного та якiсного складу лiкарського засобу та/або пакувальних матерiалiв, термiну придатностi, умов зберiгання, найменування та мiсцезнаходження виробника/заявника тощо;
помилки у графiчному зображеннi первинної, вторинної упаковок, зокрема штрих-кодi, мiсцезнаходженнi, номерi серiї, датi виробництва, термiнi придатностi тощо; орфографiчнi та/або граматичнi помилки;
помилки у специфiкацiї та/або методах контролю, зокрема цифровi, та помилки, пов'язанi з перекладом; помилки в розрахункових формулах, що виникли при скороченнi розгорнутих формул;
помилки при посиланнi на загальнi статтi та монографiї фармакопеї, нормативнi документи за умови, що основна iнформацiя роздiлу мiстить вищезазначенi посилання;
ґ) невiдповiднiсть iнформацiї (рiзночитання) у межах одного документа;
д) орфографiчнi та/або граматичнi помилки у короткiй характеристицi лiкарського засобу та iнструкцiї для медичного застосування лiкарського засобу за умови, що вони не впливають на змiст;
е) наявнiсть розбiжностей у короткiй характеристицi лiкарського засобу та iнструкцiї для медичного застосування лiкарського засобу, якi були затвердженi одночасно, щодо викладення iнформацiї, яка стосується медичного застосування лiкарського засобу; зазначення доз препарату в одиницях лiкарської форми, детальнiше викладення iнформацiї в одному з документiв;
є) внесення до затверджених пiд час реєстрацiї (перереєстрацiї) лiкарського засобу документiв назви лiкарського засобу або МНН за умови, що така iнформацiя наявна хоча б в одному з таких документiв;
ж) орфографiчнi та/або граматичнi помилки у реєстрацiйному посвiдченнi на лiкарський засiб; наявнiсть розбiжностей мiж iнформацiєю, наведеною у реєстрацiйному посвiдченнi на лiкарський засiб, та в затверджених короткiй характеристицi, та/або iнструкцiї для медичного застосування, та/або МКЯ.
Виправлення технiчної помилки затверджується наказом МОЗ.
4.2.4. Змiни, що потребують нової реєстрацiї лiкарського засобу:
4.2.4.1. Змiни активних речовин:
а) замiна активної речовини на iншу сiль, ефiр, похiдну тощо за умови, що характеристики, якi визначають спiввiдношення користь/ризик, суттєво не вiдрiзняються вiд зареєстрованих або використання iнших iзомерiв, або змiна iзомерного складу, або незначнi змiни молекулярної структури речовин бiологiчного походження, за винятком змiни у АФI або дiючiй речовинi вакцин для профiлактики грипу;
б) змiна вектора, який використовується для виробництва антигена або вихiдного матерiалу, включаючи новий головний банк клiтин вiд iншого джерела, якщо характеристики, якi визначають спiввiдношення користь/ризик, суттєво не вiдрiзняються вiд зареєстрованих;
в) новий лiганд або механiзм з'єднання для радiофармацевтичного лiкарського засобу, якщо характеристики, якi визначають спiввiдношення користь/ризик, суттєво не вiдрiзняються вiд зареєстрованих;
г) застосування iнших екстрагентiв або спiввiдношення рослинна лiкарська сировина/рослинний препарат для рослинного лiкарського засобу, якщо характеристики, якi визначають спiввiдношення користь/ризик, суттєво не вiдрiзняються вiд зареєстрованих.
4.2.4.2. Змiни сили дiї, лiкарської форми та способу застосування:
а) змiна бiодоступностi;
б) змiна фармакокiнетики;
в) змiна або введення додаткового дозування лiкарського засобу (додаткова доза);
г) змiна або додання нової лiкарської форми;
ґ) змiна або додання нового шляху введення (для парентеральних форм у зв'язку з вiдмiнностями в ефективностi та безпецi лiкарського засобу при внутрiшньоартерiальному, внутрiшньовенному, внутрiшньом'язовому та iнших шляхах уведення).
При таких змiнах заявник разом з обґрунтуванням потреби внесення змiн подає до Центру вiдповiднi роздiли матерiалiв реєстрацiйного досьє, якi обґрунтовують указанi змiни i є достатнiми для експертизи лiкарського засобу.
4.3. Експертизi пiдлягає кожна конкретна змiна навiть за умови одночасного їх внесення.
4.4. Для проведення експертизи матерiалiв про внесення змiн заявник подає до МОЗ:
для змiн типу IА, IБ або II - заяву згiдно з додатком 12 до цього Порядку;
для змiн, що потребують нової реєстрацiї, - заяву згiдно з додатком 1 до цього Порядку;
для виправлення технiчної помилки - лист у довiльнiй формi, у якому зазначенi помилка та обґрунтування її виправлення;
для технiчних змiн - лист у довiльнiй формi, у якому зазначено обґрунтування змiн.
До заяви додаються:
копiя чинного реєстрацiйного посвiдчення;
пiдтвердження затвердження запропонованих змiн уповноваженим органом країни виробника/заявника (за необхiдностi).
За результатами розгляду заяви МОЗ надсилає до Центру лист-направлення разом iз копiєю заяви для внесення змiн. Iнформацiя про подану до МОЗ заяву вноситься до електронної бази даних.
Пiсля надходження до Центру листа-направлення вiд МОЗ Центр протягом 3 робочих днiв iнформує про це заявника i надає рахунок на сплату згiдно з договором мiж заявником та Центром. Заявник здiйснює оплату та подає до Центру матерiали реєстрацiйного досьє, що обґрунтовують запропонованi змiни. Центр приймає матерiали реєстрацiйного досьє у строк, що не перевищує 3 робочi днi з дати звернення заявника.
У разi ненадання заявником матерiалiв реєстрацiйного досьє протягом 3 мiсяцiв з дати iнформування Центром заявника Центр надає МОЗ рекомендацiї щодо зняття заяви з розгляду. МОЗ протягом 5 робочих днiв розглядає наданi рекомендацiї та приймає вiдповiдне рiшення. Про прийняте рiшення МОЗ письмово повiдомляє заявника. При цьому вартiсть проведення експертних робiт заявнику не повертається. Надалi така заява може бути подана до МОЗ за процедурою, установленою цим Порядком. Вiдповiдна iнформацiя вноситься до електронної бази даних.
4.4.1. У разi внесення змiн типу IА або IБ до реєстрацiйного досьє одного лiкарського засобу заява повинна стосуватися лише однiєї змiни типу IА або IБ. У разi одночасного внесення декiлькох змiн типу IА або IБ окрема заява подається щодо кожної змiни, яка повинна мiстити посилання на iншi заяви про внесення змiн.
У разi якщо змiна типу IА призводить до iнших послiдовних змiн типу IА, одна заява може включати всi послiдовнi змiни разом з описом вiдповiдностей мiж такими послiдовними змiнами типу IА.
У разi якщо змiна типу IБ призводить до iнших послiдовних змiн типу IА або IБ, одна заява може включати всi послiдовнi змiни разом з описом вiдповiдностей мiж такими послiдовними змiнами типу IА або IБ.
Якщо змiни потребують послiдовної перевiрки короткої характеристики, iнструкцiї для медичного застосування лiкарського засобу, МКЯ, що мiстить маркування первинної, вторинної (за наявностi) упаковок, це вважається частиною змiн.
4.4.2. У разi внесення значних змiн типу II заява повинна стосуватися лише однiєї змiни типу II. Якщо необхiдно внести декiлька змiн типу II, окрема заява повинна подаватися щодо кожної змiни; кожна така заява повинна мiстити посилання на iншу заяву.
У разi якщо змiна типу II призводить до iнших послiдовних змiн типу II, одна заява може включати всi послiдовнi змiни разом з описом вiдповiдностей мiж такими послiдовними змiнами типу II.
Якщо змiни потребують послiдовної перевiрки короткої характеристики, iнструкцiї для медичного застосування, МКЯ, що мiстить маркування первинної, вторинної (за наявностi) упаковок, це вважається частиною змiн.
4.4.3. У разi внесення технiчних змiн, що стосуються змiни найменування виробника та/або заявника, що вносяться заявником для приведення у вiдповiднiсть з нормативно-правовими актами України, зокрема Законом України "Про акцiонернi товариства", або стосуються, наприклад, змiни контактної iнформацiї, що призводить до необхiдностi оновлення iнформацiї у вiдповiдних роздiлах матерiалiв реєстрацiйного досьє, такi змiни не потребують проведення експертизи експертами з питань реєстрацiї Центру. Перевiрка матерiалiв реєстрацiйного досьє здiйснюється Центром на безоплатнiй основi та у строк до 10 робочих днiв з дати надходження листа-направлення вiд МОЗ.
4.4.4. У випадку вiдсутностi в додатку 11 до цього Порядку класифiкацiї змiн, що вносяться до матерiалiв реєстрацiйного досьє пiд час дiї реєстрацiйного посвiдчення, ця змiна зазначається як пiдпункт "Iншi змiни" у вiдповiдному пунктi додатка 12 до цього Порядку.
Данi змiни за класифiкацiєю можуть бути як змiни типу IА, IБ, так i змiни II типу.
До змiн типу IА вiдносять:
а) змiни виключно адмiнiстративного характеру:
змiна найменування та/або мiсцезнаходження заявника (власника реєстрацiйного посвiдчення);
змiна найменування та/або мiсцезнаходження виробника готового лiкарського засобу;
змiна найменування та/або мiсцезнаходження виробника субстанцiї;
б) виключення будь-якої виробничої дiльницi для таких стадiй виробництва готового лiкарського засобу: виробництво АФI або дiючої речовини, виробництво промiжного продукту чи готового лiкарського засобу, пакування, випуск серiї, контроль серiї;
в) змiни у МКЯ АФI або дiючої речовини або вихiдної сировини/промiжних продуктiв або реагентiв, що використовуються у виробничому процесi, за умови, що не змiнюється сама методика (наприклад, змiнюється довжина хроматографiчної колонки, але не її тип), проведена валiдацiя змiненого методу контролю, яка показує вiдповiднiсть попередньому методу, та не змiнюються межi, встановленi у специфiкацiї;
г) змiни, що вносяться у зв'язку зi змiнами у ДФУ, або Європейськiй фармакопеї, або iншiй фармакопеї (якщо субстанцiя не описана у ДФУ чи Європейськiй фармакопеї), або приведення у вiдповiднiсть до цих фармакопей;
ґ) змiни в упаковцi лiкарського засобу, що не контактує безпосередньо з лiкарським засобом та не впливає на доставку, застосування, безпеку або стабiльнiсть лiкарського засобу;
д) змiни у специфiкацiях на лiкарський засiб, АФI або дiючу речовину, вихiднi матерiали, промiжнi продукти, допомiжнi речовини, що призводять до звуження меж, за умови, що новi параметри залишаються у рамках заявленого дiапазону та не є результатом непередбачених обставин у процесi виробництва.
До змiн типу II вiдносять:
а) змiни, пов'язанi з введенням нового показання до застосування або змiнами вже iснуючого;
б) значнi змiни у короткiй характеристицi лiкарського засобу, зокрема, на пiдставi нових вiдомостей щодо якостi, результатiв доклiнiчних дослiджень та клiнiчних випробувань, нових повiдомлень про безпеку лiкарського засобу, включаючи термiновi змiни, що стосуються безпеки;
в) змiни, пов'язанi зi змiнами поза дiапазоном затверджених специфiкацiй, меж та критерiїв прийнятностi, якщо це не пов'язано iз приведенням у вiдповiднiсть до вимог ДФУ чи Європейської фармакопеї;
г) змiни, що стосуються суттєвих змiн у виробничому процесi, складi лiкарського засобу, специфiкацiях, складi домiшок у АФI або дiючiй речовинi чи готового лiкарського засобу, якi можуть мати iстотний вплив на його якiсть, безпеку та ефективнiсть;
ґ) змiни, пов'язанi зi змiнами у виробничому процесi або дiльницi для виробництва АФI або дiючої речовини бiологiчного походження;
д) змiни, пов'язанi з введенням нового проектного простору або розширенням затвердженого, який був розроблений вiдповiдно до Європейських та мiжнародних наукових керiвництв;
е) змiни заявника, крiм одночасних змiн заявника та мiсцезнаходження виробництва.
До змiн типу IБ вiдносять незначнi змiни, якi не можуть бути змiнами типу IА та типу II i не потребують нової реєстрацiї.
4.5. Експертиза пропозицiй щодо внесення змiн до матерiалiв реєстрацiйного досьє проводиться у Центрi на пiдставi листа-направлення МОЗ пiсля отримання матерiалiв реєстрацiйного досьє, що пiдтверджують запропонованi змiни, та оплати її вартостi, установленої договором мiж заявником та Центром.
4.6. При внесеннi змiн типу IА, IБ до Центру на експертизу подаються матерiали згiдно з додатком 11 до цього Порядку.
При внесеннi змiн II-го типу заявник повинен надати Центру:
матерiали, якi обґрунтовують унесення змiн;
вiдповiднi матерiали з поправками, унесеними вiдповiдно до заяви;
доповненi або оновленi iснуючi експертнi звiти (огляди, висновки) з урахуванням змiн (за необхiдностi);
якщо змiни призводять до змiн у МКЯ, що мiстять маркування первинної, вторинної (за наявностi) упаковок лiкарського засобу, iнструкцiї для медичного застосування, примiрники цих документiв для затвердження, викладенi вiдповiдно до вимог роздiлiв XVI та XVIII цього Порядку та додатка 10 до цього Порядку.
У разi внесення змiн в iнструкцiю для медичного застосування, якi не стосуються та не впливають на клiнiчну iнформацiю (назва лiкарського засобу, склад, лiкарська форма, основнi фiзико-хiмiчнi властивостi, виробник, мiсцезнаходження, умови зберiгання, термiн придатностi тощо), у складi iнших матерiалiв реєстрацiйного досьє разом iз заявою заявник подає змiни до iнструкцiї для медичного застосування лiкарського засобу за формою, наведеною у додатку 13 до цього Порядку.
За бажанням заявника матерiали реєстрацiйного досьє можуть подаватися у виглядi змiн до певних роздiлiв реєстрацiйного досьє у форматi ЗТД.
4.7. Експертиза матерiалiв про внесення змiн складається з таких етапiв:
попередньої експертизи матерiалiв реєстрацiйного досьє, метою якої є перевiрка їх вiдповiдностi заявленому типу змiн, яка триває 5 робочих днiв з дати їх надходження до Центру;
спецiалiзованої експертизи матерiалiв реєстрацiйного досьє про внесення змiн.
Розгляд виправлення технiчних помилок здiйснюється у рамках попередньої експертизи (протягом 20 днiв з дати надання заявником листа-звернення та необхiдних документiв, що їх обґрунтовують) шляхом порiвняння виправлених матерiалiв реєстрацiйного досьє з матерiалами оригiнального реєстрацiйного досьє заявника.
4.8. У разi вiдповiдностi матерiалiв реєстрацiйного досьє лiкарського засобу заявленому типу змiн, встановлених за результатами попередньої експертизи матерiалiв реєстрацiйного досьє, вони направляються на спецiалiзовану експертизу, а у випадку виправлення технiчних помилок та внесення технiчних змiн Центром надаються рекомендацiї МОЗ щодо їх затвердження, про що Центр письмово повiдомляє заявника та вносить вiдповiдну iнформацiю до електронної бази даних.
При процедурi виправлення технiчних помилок та внесеннi технiчних змiн спецiалiзована експертиза матерiалiв реєстрацiйного досьє не проводиться.
4.9. У разi невiдповiдностi матерiалiв реєстрацiйного досьє лiкарського засобу заявленому типу змiн, встановлених за результатами попередньої експертизи матерiалiв реєстрацiйного досьє, Центр повiдомляє про це заявника письмово i надає вiдповiдне обґрунтування з посиланням на номери роздiлiв, глав, статей, пунктiв цього Порядку або одноразово запитує у заявника додатковi данi та/або iнформацiю, необхiднi для забезпечення вiдповiдностi наданих матерiалiв реєстрацiйного досьє вимогам. Вiдповiдна iнформацiя вноситься до електронної бази даних.
Заявник має надати додатковi данi та/або iнформацiю згiдно iз зауваженнями Центру, або листа з обґрунтуванням термiнiв, необхiдних для їх доопрацювання, у строк до 30 робочих днiв. Час, потрiбний для доопрацювання, не входить до строку проведення експертизи. Центр має прийняти доопрацьованi матерiали протягом 3 робочих днiв пiсля звернення заявника. Вiдповiдна iнформацiя вноситься до електронної бази даних.
Якщо заявник не надає Центру додаткових даних та/або iнформацiї, а також якщо наданi заявником додатковi данi та/або iнформацiя не забезпечують вiдповiдностi матерiалiв реєстрацiйного досьє встановленим вимогам, то Центр надає МОЗ рекомендацiї щодо вiдмови у внесеннi змiн до матерiалiв реєстрацiйного досьє лiкарського засобу.
Для змiн типу IА додатковi матерiали та/або iнформацiя у заявника не запитуються. У разi невiдповiдностi матерiалiв реєстрацiйного досьє лiкарського засобу заявленому типу змiн, вимоги до яких викладено в додатку 11 до цього Порядку, Центр надає МОЗ рекомендацiї щодо вiдмови у внесеннi змiн до матерiалiв реєстрацiйного досьє лiкарського засобу.
МОЗ у строк до 10 робочих днiв розглядає наданi рекомендацiї та приймає вiдповiдне рiшення, що затверджується наказом. Про прийняте рiшення МОЗ письмово повiдомляє заявника. При цьому вартiсть проведення експертних робiт заявнику не повертається. Вiдповiдна iнформацiя вноситься до електронної бази даних.
Надалi на бажання заявника матерiали про внесення змiн до матерiалiв реєстрацiйного досьє протягом дiї реєстрацiйного посвiдчення з урахуванням зауважень Центру подаються в установленому порядку.
4.10. Пiд час проведення спецiалiзованої експертизи матерiалiв реєстрацiйного досьє про внесення змiн з метою одержання додаткових даних щодо впливу змiн на ефективнiсть, безпеку та якiсть лiкарського засобу Центр може одноразово запитати у заявника додатковi данi та/або iнформацiю з посиланням на номери роздiлiв, глав, статей, пунктiв цього Порядку. Час, потрiбний для їх пiдготовки, не входить до строку проведення експертних робiт.
Пiсля отримання запитаних додаткових даних та/або iнформацiї Центр здiйснює їх експертизу у строк, що не перевищує 30 календарних днiв вiд дати отримання Центром додаткових матерiалiв.
У разi невiдповiдностi наданих матерiалiв встановленим вимогам Центр надає МОЗ рекомендацiї щодо вiдмови у внесеннi змiн. Вiдповiдна iнформацiя вноситься до електронної бази даних. МОЗ у строк до 10 робочих днiв розглядає наданi рекомендацiї та приймає вiдповiдне рiшення, що затверджується наказом МОЗ. Про прийняте рiшення МОЗ письмово повiдомляє заявнику.
Якщо заявник протягом 60 робочих днiв не надсилає запитаних додаткових даних та/або iнформацiї або листа з обґрунтуванням строкiв, необхiдних для їх пiдготовки, або надає їх у неповному обсязi, Центр в установленому порядку надає МОЗ рекомендацiї щодо вiдмови у внесеннi змiн. Вiдповiдна iнформацiя вноситься до електронної бази даних.
Для змiн типу IA додатковi матерiали та/або iнформацiя у заявника не запитуються. У разi невiдповiдностi матерiалiв реєстрацiйного досьє лiкарського засобу заявленому типу змiн, вимоги до яких викладено в додатку 11 до цього Порядку, Центр надає МОЗ рекомендацiї щодо вiдмови у внесеннi змiн до матерiалiв реєстрацiйного досьє лiкарського засобу.
МОЗ у строк до 10 робочих днiв розглядає наданi рекомендацiї та приймає вiдповiдне рiшення, що затверджується наказом. Про прийняте рiшення МОЗ письмово повiдомляє заявника. При цьому вартiсть проведення експертних робiт заявнику не повертається. Надалi на бажання заявника матерiали про внесення змiн можуть бути поданi в установленому порядку.
4.11. За результатами експертизи Центр надає рекомендацiї та висновки до МОЗ щодо внесення змiн до матерiалiв реєстрацiйного досьє або змiн, що потребують нової реєстрацiї лiкарського засобу, враховуючи при цьому строк, зазначений заявником у заявi, протягом якого заявленi змiни мають бути введенi в дiю, що вiдображається в наказi МОЗ щодо затвердження вiдповiдних змiн. Вiдповiдна iнформацiя вноситься до електронної бази даних.
Про прийняте рiшення МОЗ письмово повiдомляє заявника.
У випадку, якщо змiни стосуються iнструкцiї для медичного застосування лiкарського засобу, МКЯ, Центр також рекомендує МОЗ до затвердження оновлену iнструкцiю для медичного застосування або змiни до неї, змiни до МКЯ, що включають текст, що наводиться у маркуваннi (за необхiдностi).
На пiдставi поданих Центром висновкiв та рекомендацiй МОЗ у мiсячний строк пiсля їх отримання приймає рiшення про внесення змiн або про вiдмову в них i розмiщує таке рiшення на сайтi МОЗ у виглядi наказу МОЗ.
Пiсля пiдписання МОЗ наказу щодо внесення змiн видача затверджених МОЗ примiрникiв документiв на лiкарський засiб, що пiдтверджують факт затвердження змiни, здiйснюється МОЗ за принципом "єдиного вiкна".
4.12. У разi незгоди з рекомендацiями Центру за результатами експертизи щодо змiн, якi потребують нової реєстрацiї, заявник може оскаржити це рiшення до МОЗ протягом 30 робочих днiв з дати його одержання. Вiдповiднi матерiали для обґрунтування оскарження мають бути наданi заявником до МОЗ протягом 30 робочих днiв пiсля подання такого оскарження.
Центр за направленням МОЗ здiйснює експертизу наданих заявником матерiалiв у строк до 60 робочих днiв з дати їх одержання та надає вiдповiднi обґрунтованi рекомендацiї МОЗ. МОЗ у мiсячний строк приймає вiдповiдне рiшення, про що iнформує письмово заявника.
V. СТРОКИ ПРОВЕДЕННЯ ЕКСПЕРТИЗИ
Для експертизи встановлюються такi строки її проведення:
5.1. Не бiльше 210 робочих днiв, починаючи з дати офiцiйного надходження до Центру матерiалiв реєстрацiйного досьє на державну реєстрацiю, триває експертиза матерiалiв щодо лiкарського засобу, який подається на державну реєстрацiю за повною i незалежною заявою (автономною заявою), а також щодо МIБП, поданого на державну реєстрацiю.
Не бiльше 90 робочих днiв, починаючи з дати офiцiйного надходження до Центру матерiалiв реєстрацiйного досьє, триває експертиза матерiалiв щодо лiкарських засобiв, зазначених у пiдпунктах 6.10.1 та 6.10.2 пункту 6.10 роздiлу VI цього Порядку.
5.2. Не бiльше 90 робочих днiв, починаючи з дати офiцiйного надходження до Центру матерiалiв реєстрацiйного досьє, триває експертиза матерiалiв щодо:
лiкарських засобiв, якi подаються на державну реєстрацiю вiдповiдно до iнших типiв заяв, визначених у додатку 1 до цього Порядку;
АФI або дiючих речовин;
лiкарських засобiв, що подаються на державну перереєстрацiю;
традицiйних лiкарських засобiв та лiкарських засобiв, що виробляються згiдно iз затвердженими прописами;
змiн, що потребують нової реєстрацiї.
5.3. Не бiльше 60 робочих днiв пiсля надходження до Центру вiдповiдних матерiалiв реєстрацiйного досьє триває експертиза матерiалiв про внесення змiн до матерiалiв реєстрацiйного досьє.
Цей перiод може бути скорочений через термiновiсть питання, якщо змiни стосуються безпеки застосування лiкарського засобу.
5.4. Датою завершення експертизи матерiалiв реєстрацiйного досьє, зазначених у пунктах 5.1 - 5.3 цього роздiлу, вважається дата пiдписання керiвником Центру висновкiв експертiв з питань реєстрацiї за результатами проведеної експертизи, а саме:
умотивованих висновкiв щодо ефективностi, безпеки та якостi лiкарського засобу та рекомендацiй його до державної реєстрацiї (перереєстрацiї);
рекомендацiй щодо вiдмови у державнiй реєстрацiї (перереєстрацiї);
висновкiв щодо внесення змiн до матерiалiв реєстрацiйного досьє чи нової реєстрацiї лiкарського засобу в установленому порядку;
рекомендацiй щодо вiдмови у внесеннi змiн до матерiалiв реєстрацiйного досьє протягом дiї реєстрацiйного посвiдчення на лiкарськiй засiб.
Висновки та рекомендацiї оформлюються Центром у виглядi перелiкiв лiкарських засобiв, рекомендованих до реєстрацiї, перереєстрацiї, внесення змiн у реєстрацiйнi матерiали та внесення до Державного реєстру лiкарських засобiв, до яких додаються експертнi висновки щодо ефективностi, безпеки та якостi лiкарського засобу та примiрники iнструкцiй для медичного застосування лiкарського засобу, фармакопейних статей або МКЯ, що мiстять маркування первинної, вторинної (за наявностi) упаковок, iз штампом Центру "контрольний примiрник"; перелiкiв лiкарських засобiв, не рекомендованих до реєстрацiї, перереєстрацiї, внесення змiн у реєстрацiйнi матерiали разом iз експертними висновками. Цi матерiали направляються до МОЗ для прийняття рiшення.
Одночасно з направленням до МОЗ перелiкiв лiкарських засобiв, рекомендованих до реєстрацiї, перереєстрацiї, внесення змiн у реєстрацiйнi матерiали та внесення до Державного реєстру лiкарських засобiв та лiкарських засобiв, яким рекомендовано вiдмовити у реєстрацiї, перереєстрацiї, внесеннi змiн у реєстрацiйнi матерiали, зазначенi перелiки розмiщуються Центром на своєму сайтi.
Зберiгання матерiалiв реєстрацiйного досьє, наведених у цьому Порядку, здiйснюється Центром.
Видача документiв заявнику, зокрема iнструкцiї для медичного застосування або змiн до неї, короткої характеристики (за необхiдностi), фармакопейної статтi або МКЯ лiкарського засобу або змiн до них, що мiстять маркування первинної, вторинної (за наявностi) упаковок, здiйснюється МОЗ за принципом "єдиного вiкна".
5.5. До строкiв експертних робiт, зазначених у пунктах 5.1 - 5.3 цього Порядку, не входить час, коли матерiали були на доопрацюваннi в заявника, час, необхiдний на отримання вiдповiдей вiд третiх осiб (у т. ч. - уповноважених органiв України та/або iнших країн) на запити Центру, пов'язанi iз проведенням експертизи, а також час проведення додаткових вивчень (випробувань).
VI. ОСНОВНI ВИМОГИ ДО МАТЕРIАЛIВ РЕЄСТРАЦIЙНОГО ДОСЬЄ
6.1. Матерiали реєстрацiйного досьє, що надаються на експертизу, повиннi мiстити:
6.1.1 для державної реєстрацiї - iнформацiю, яка зазначена у роздiлах IX та X цього Порядку (готовi лiкарськi форми, продукцiя in bulk, продукцiя iз in bulk), роздiлi XIII цього Порядку (традицiйнi лiкарськi засоби рослинного походження), роздiлi XIV цього Порядку (лiкарськi засоби, що виробляються за затвердженими прописами) або роздiлi XI цього Порядку (дiючi речовини або АФI, зокрема у формах пелет, гранул та iнших формах випуску);
6.1.2 для державної перереєстрацiї - iнформацiю, яка зазначена у додатку 7 або додатку 8 до цього Порядку (традицiйнi лiкарськi засоби або лiкарськi засоби, якi виробляються згiдно iз затвердженим прописом);
6.1.3 для внесення змiн до матерiалiв реєстрацiйного досьє - iнформацiю, яка зазначена у додатку 11 до цього Порядку, або iнформацiю, необхiдну для експертизи для внесення змiн II типу з урахуванням вимог пункту 4.6 роздiлу IV цього Порядку, та змiн, що потребують нової реєстрацiї з урахуванням вимог додатка 5 до цього Порядку та роздiлiв IX та X цього Порядку.
При пiдготовцi матерiалiв реєстрацiйного досьє заявник має керуватися вимогами, викладеними у роздiлах VII та VIII цього Порядку (тiльки при перереєстрацiї), або при пiдготовцi матерiалiв реєстрацiйного досьє у форматi ЗТД - вимогами роздiлiв IX та X цього Порядку, з урахуванням вимог роздiлiв XVI, XVII, XVIII цього Порядку та додаткiв 10, 14 до цього Порядку, у разi традицiйних лiкарських засобiв рослинного походження та лiкарських засобiв, що виробляються згiдно iз затвердженими прописами, - роздiлiв XIII та XIV цього Порядку, гомеопатичних лiкарських засобiв - роздiлу XII цього Порядку.
Для перереєстрацiї лiкарських засобiв надається комплект матерiалiв реєстрацiйного досьє залежно вiд типу лiкарського засобу вiдповiдно до додатка 7 або 8 до цього Порядку.
Для державної реєстрацiї лiкарських засобiв, на якi вiдповiдно до чинного законодавства України видано патент, заявник подає копiю патенту або лiцензiї, якою дозволяється виробництво та продаж зареєстрованого лiкарського засобу, а також документ, що пiдтверджує чиннiсть патенту в Українi. Заявник подає лист, в якому вказується, що права третьої сторони, захищенi патентом або переданi за лiцензiєю, не порушуються у зв'язку з реєстрацiєю лiкарського засобу, за формою, наведеною у додатку 15 до цього Порядку.
У разi реєстрацiї АФI або дiючих речовин можливе їх використання як лiкарських засобiв в упаковцi in bulk, якщо АФI або дiючi речовини частково пройшли технологiчнi стадiї їх переробки у готову лiкарську форму (зокрема, у формах: грануляту, пелет, гранул, покритих оболонкою, iнших формах випуску). У такому випадку визначається обсяг додаткових матерiалiв у форматi ЗТД (додаток 5 до цього Порядку) для пiдтвердження властивостей, наданих АФI або дiючим речовинам внаслiдок зазначеної часткової технологiчної переробки (пролонгована або вiдтермiнована дiя, стiйкiсть до дiї шлункового соку тощо).
6.2. При реєстрацiї готового лiкарського засобу специфiкацiї на дiючу речовину (роздiли реєстрацiйного досьє 3.2.S.4 додатка 5 до цього Порядку) можуть мiстити додатковi показники щодо технологiчних параметрiв АФI або дiючої речовини, обґрунтованi при фармацевтичнiй розробцi (роздiли реєстрацiйного досьє 3.2.S.2 додатка 5 до цього Порядку), та доповнюють показники специфiкацiй, що мiстяться у МКЯ на АФI або дiючу речовину та/або у сертифiкатi якостi, виданому виробником.
6.3. Вимоги до якостi лiкарського засобу повиннi бути не нижче вiд встановлених у ДФУ та/або Європейськiй фармакопеї.
У разi вiдсутностi вiдповiдної монографiї на готовий лiкарськiй засiб у ДФУ та/або Європейськiй фармакопеї вимоги до якостi лiкарського засобу повиннi бути не нижче вiд встановлених в iнших провiдних фармакопеях свiту (Британська фармакопея, фармакопея США, фармакопея Японiї).
У разi вiдсутностi вiдповiдної монографiї на готовий лiкарськiй засiб у вищезазначених фармакопеях вимоги до якостi генеричного лiкарського засобу повиннi встановлюватися виробником та вiдповiдним чином обґрунтовуватися на пiдставi доведення взаємозамiнностi з референтним препаратом.
У разi внесення змiн до загальних або окремих статей ДФУ та/або Європейської фармакопеї та/або Британської фармакопеї та/або фармакопеї США та/або фармакопеї Японiї заявник має протягом 6 мiсяцiв з дати введення в дiю цих змiн привести у вiдповiднiсть до них дiючi методи контролю якостi зареєстрованого лiкарського засобу.
6.4. Вiд заявника не вимагається надання результатiв власних токсикологiчних та фармакологiчних випробувань або результатiв клiнiчних випробувань, якщо вiн може надати один з таких доказiв:
6.4.1. Лiкарський засiб є генериком до референтного лiкарського засобу i заявник вже зареєстрованого референтного лiкарського засобу згоден з тим, аби при вивченнi заяви на генерик були використанi данi фармакологiчних, токсикологiчних та/або клiнiчних випробувань, якi мiстяться в реєстрацiйних матерiалах на референтний лiкарський засiб (глава 2 роздiлу X цього Порядку).
6.4.2. Лiкарський засiб є генериком до референтного лiкарського засобу, що доведено дослiдженнями, проведеними вiдповiдно до роздiлу XX цього Порядку.
Дослiдження бiодоступностi не вимагатимуться вiд заявника, якщо вiн може довести вiдповiднiсть генеричного лiкарського засобу критерiям, зазначеним у роздiлi XX цього Порядку.
У разi коли при реєстрацiї генеричного лiкарського засобу, еквiвалентнiсть якого має бути доведена фармакокiнетичними дослiдженнями (бiоеквiвалентнiсть), результати цих дослiджень ще не доступнi, а представленi дослiдження еквiвалентностi проведено iз застосуванням iнших методiв, нiж тi, що передбаченi роздiлом XX цього Порядку, що належним чином обґрунтовано заявником, то реєстрацiйне досьє може бути прийняте з урахуванням певних зобов'язань. Цi зобов'язання повиннi включати програму дослiджень бiоеквiвалентностi генеричного лiкарського засобу до референтного згiдно з роздiлом XX цього Порядку, якi мають бути проведенi протягом двох рокiв з дати отримання заявником реєстрацiйного посвiдчення, та результати дослiджень мають бути внесенi до матерiалiв реєстрацiйного досьє в установленому порядку.
При цьому iнструкцiя для медичного застосування лiкарського засобу генеричного лiкарського засобу має мiстити iнформацiю, iдентичну до тiєї, яка наведена в iнструкцiї для медичного застосування референтного лiкарського засобу.
Будь-якi вiдмiнностi iнструкцiї для медичного застосування генеричного лiкарського засобу мають обґрунтовуватись результатами, отриманими при багатоцентрових клiнiчних дослiдженнях (глава 2 роздiлу X цього Порядку).
6.4.3. Медичне застосування АФI або дiючої речовини, або речовин, якi входять до складу лiкарського засобу, добре вивчене (абзац двадцять третiй роздiлу II цього Порядку), визнано їх ефективнiсть та задовiльний ступiнь безпеки (шляхом надання докладних бiблiографiчних посилань на опублiкованi науковi данi) (глава 1 роздiлу X цього Порядку).
Якщо надано бiблiографiчнi посилання на опублiкованi науковi данi, вони викладаються з урахуванням вимог, наведених у роздiлах IX та X цього Порядку в обсягах, якi дозволяють заявнику не надавати результатiв власних токсикологiчних та фармакологiчних випробувань або результатiв клiнiчних випробувань.
6.5. Якщо лiкарськi засоби мiстять у своєму складi вiдомi АФI або дiючi речовини, якi ранiше не застосовувались у даному сполученнi з терапевтичною метою, то необхiдно надати результати токсикологiчних, фармакологiчних або клiнiчних випробувань, що належать до цього сполучення; при цьому немає необхiдностi надавати посилання, якi стосуються до кожної складової (глава 4 роздiлу X цього Порядку).
Це не стосується випадкiв, коли комбiнованi лiкарськi засоби мiстять вiдомi АФI або дiючi речовини, якi ранiше використовувались у медичнiй практицi як окремi лiкарськi засоби у тих самих лiкарськiй формi, дозах та вiдповiдному спiввiдношеннi для досягнення тiєї самої терапевтичної мети. При цьому необхiдно надати результати дослiджень, що засвiдчать як на стадiї виробництва, так i протягом термiну зберiгання комбiнованого лiкарського засобу вiдсутнiсть мiж складовими будь-якої взаємодiї, яка може вплинути на ефективнiсть та безпеку лiкарського засобу.
6.6. У випадку реєстрацiї подiбних бiологiчних лiкарських засобiв (бiосимiлярiв), якi не можуть вважатись генериками через специфiчнiсть процесу виробництва, застосовуваної сировини, особливостей молекулярної будови та терапевтичної дiї, крiм модулiв 1, 2, 3 необхiдно надати додатковi данi, що пiдтверджують належний рiвень безпеки (токсикологiчнi чи iншi доклiнiчнi вивчення) та ефективностi (клiнiчнi випробування) (глава 3 роздiлу X цього Порядку).
6.7. Якщо лiкарськi засоби є гомеопатичними лiкарськими засобами, якi вiдповiдають умовам роздiлу XII цього Порядку, чи традицiйними лiкарськими засобами рослинного походження, якi вiдповiдають умовам роздiлу XIII цього Порядку, або лiкарськими засобами, що виробляються згiдно iз затвердженими МОЗ прописами, якi вiдповiдають умовам роздiлу XIV цього Порядку, вiд заявника не вимагаються науковi клiнiчнi данi. При цьому матерiали щодо якостi лiкарського засобу повиннi бути наданi в повному обсязi, а також у разi виникнення сумнiвiв щодо його безпеки на запит Центру повиннi надаватися данi, необхiднi для експертизи безпеки лiкарського засобу (глави 10, 11 роздiлу X цього Порядку).
6.8. Матерiали щодо державної реєстрацiї джерела радiонуклiдiв повиннi також мiстити таку iнформацiю та характеристики (вимоги викладено у главi 9 роздiлу X цього Порядку):
загальний опис системи разом з докладним описом компонентiв системи, якi можуть вплинути на склад та якiсть лiкарського засобу iз вторинних радiонуклiдiв;
якiснi та кiлькiснi характеристики елюату або сублiмату.
6.9. Обсяг матерiалiв реєстрацiйного досьє на лiкарськi засоби, що подаються на державну реєстрацiю, визначається типом заяви (додаток 1 або додаток 2 до цього Порядку). Перелiк документiв для проведення експертизи матерiалiв для державної реєстрацiї АФI або дiючої речовини, медичних газiв визначено в додатку 6 до цього Порядку.
6.10. З метою надання необхiдної медичної допомоги населенню при лiкуваннi соцiально небезпечних хвороб (туберкульоз, ВIЛ/СНIД, вiруснi гепатити, а також рiдкiснi захворювання) при реєстрацiї оригiнальних лiкарських засобiв та лiкарських засобiв, що пройшли процедуру преквалiфiкацiї ВООЗ та включенi до перелiку ВООЗ преквалiфiкованих лiкарських засобiв, можуть бути застосованi окремi вимоги до матерiалiв реєстрацiйного досьє на такi лiкарськi засоби:
6.10.1. Оригiнальний (iнновацiйний) лiкарський засiб для лiкування соцiально небезпечних хвороб (туберкульоз, ВIЛ/СНIД, вiруснi гепатити, а також лiкарський засiб з оригiнальною молекулою для лiкування рiдкiсних захворювань) був зареєстрований в країнi, регуляторнi органи якої застосовують високi стандарти до якостi, що вiдповiдають стандартам, рекомендованим ВООЗ, зокрема: Адмiнiстрацiя з харчових продуктiв та лiкарських засобiв США (FDA); Європейське агентство з медичних продуктiв (EMA) (за централiзованою процедурою); Агенцiя з терапевтичних продуктiв Швейцарiї (Swissmedic); Агентство з лiкарських засобiв та продуктiв медичного призначення Японiї (PMDA); Агентство з регулювання лiкарських засобiв та продуктiв медичного призначення Великобританiї (MHRA); Австралiйська адмiнiстрацiя лiкарських засобiв (TGA). Крiм того, необхiдно надати:
а) офiцiйний звiт з оцiнки лiкарського засобу, складений регуляторним органом, завiрений печаткою заявника (для фiзичної особи - за наявностi)/представника заявника;
б) письмове пiдтвердження вiд заявника, що заявлений на реєстрацiю лiкарський засiб вiдповiдає лiкарському засобу, що зареєстрований в країнi, регуляторний орган якої застосовує високi стандарти до якостi, що вiдповiдають стандартам, рекомендованим ВООЗ.
6.10.2. Лiкарський засiб для лiкування туберкульозу або ВIЛ/СНIДу пройшов процедуру преквалiфiкацiї та включений до перелiку ВООЗ преквалiфiкованих лiкарських засобiв (далi - перелiк ВООЗ). При цьому поданий на реєстрацiю лiкарський засiб має вироблятися на виробничiй дiльницi, що пройшла iнспектування ВООЗ у рамках програми преквалiфiкацiї та зазначена у перелiку ВООЗ; вид та розмiр упаковки заявленого до реєстрацiї лiкарського засобу зазначено в перелiку ВООЗ. Крiм того, необхiдно надати:
а) офiцiйний звiт ВООЗ з оцiнки препарату (WHOPAS(s)), завiрений печаткою заявника/представника заявника;
б) офiцiйний звiт ВООЗ з iнспектування виробникiв та вiддiлiв клiнiчного тестування (WHOPIR(s)), завiрений печаткою заявника/представника заявника;
в) офiцiйне пiдтвердження вiд ВООЗ стосовно чинностi вищезазначених документiв на дату проведення державної реєстрацiї лiкарського засобу в Українi, а також зобов'язання щодо своєчасного iнформування Центру у разi анулювання або внесення будь-яких змiн до статусу преквалiфiкованого лiкарського засобу;
г) письмове пiдтвердження вiд заявника, що заявлений на реєстрацiю лiкарський засiб вiдповiдає лiкарському засобу, що пройшов преквалiфiкацiю, та внесений до офiцiйного перелiку ВООЗ на пiдставi документiв, зазначених у пiдпунктах "а" та "б" цього пункту. При цьому лiкарський засiб може реєструватися в Українi пiд назвою, що вiдрiзняється вiд назви, пiд якою вiн зареєстрований в iншiй країнi або зазначений у перелiку ВООЗ.
6.10.3. У таких випадках заявник може подавати iнформацiю, що мiститься в модулi 1 додатка 5 до цього Порядку (з перекладом на українську або росiйську мову) та модулi 2 додатка 5 до цього Порядку (з перекладом на українську або росiйську мову за необхiдностi), на паперовому носiї. Модулi 3, 4 та 5 матерiалiв реєстрацiйного досьє додатка 5 до цього Порядку заявник може подавати в електронному виглядi, а також МКЯ, що мiстить маркування первинної, вторинної (за наявностi) упаковок, iнструкцiю для медичного застосування лiкарського засобу, а за бажанням заявника - коротку характеристику лiкарського засобу.
При реєстрацiї лiкарських засобiв, визначених у пiдпунктах 6.10.1 та 6.10.2 пункту 6.10 цього роздiлу, експертиза реєстрацiйних матерiалiв здiйснюється позачергово та без сплати вартостi експертних робiт. Передреєстрацiйний контроль за показниками якостi таких лiкарських засобiв може проводитися за процедурою "Lot Release" (шляхом експертизи матерiалiв контролю якостi серiї лiкарського засобу (протоколи контролю якостi серiї, аналiтичнi звiти), наданих виробником) (далi - "Lot Release").
6.11. Матерiали реєстрацiйного досьє подаються до Центру вiдповiдно до типу заяви у 3-х примiрниках. За погодженням з Центром заявник може подавати окремi частини реєстрацiйного досьє на електронному носiї (модуль 5 додатка 5 до цього Порядку).
Матерiали реєстрацiйного досьє подаються українською, або росiйською, або англiйською мовою. У разi подання реєстрацiйного досьє англiйською мовою заявник має надати також переклад на українську або росiйську мову модуля 1 додатка 5 до цього Порядку, а на вимогу Центру - модуля 2 додатка 5 до цього Порядку, а також методи контролю якостi готового лiкарського засобу.
Матерiали реєстрацiйного досьє, що надаються для експертизи пiд час державної реєстрацiї АФI або дiючої речовини, подаються українською, або росiйською, або англiйською мовою. У разi подання реєстрацiйного досьє англiйською мовою заявник має надати також переклад на українську або росiйську мову модуля 1 додатка 6 до цього Порядку, а на вимогу Центру - модуля 2 додатка 6 до цього Порядку, а також методи контролю якостi готового лiкарського засобу.
Переклад реєстрацiйних матерiалiв має бути засвiдчений пiдписом особи, яка його здiйснила, iз зазначенням прiзвища, iменi, по батьковi та посади такої особи, а також скрiплений печаткою заявника.
6.12. Перелiк документiв для проведення експертизи матерiалiв для державної перереєстрацiї лiкарського засобу визначено в додатку 7 до цього Порядку або за бажанням заявника - у форматi ЗТД (додаток 5 до цього Порядку), до якого мають додаватися:
регулярно оновлюваний звiт з безпеки лiкарського засобу та/або узагальнюючий звiт;
перелiк одержаних за останнi 5 рокiв в Українi рекламацiй на лiкарський засiб (у хронологiчному порядку) з докладним аналiзом причин, що були пiдставами для видачi рекламацiй, та заходiв, ужитих заявником для запобiгання їм у майбутньому;
перелiк гарантiй заявника та зобов'язань, наданих пiсля реєстрацiї/перереєстрацiї (у хронологiчному порядку), iз зазначенням характеру, стану виконання (рекомендацiї щодо пiсляреєстрацiйних дослiджень, усунення будь-яких недолiкiв, що надавалися контрольними та iншими органами тощо);
для генерикiв, у яких на час реєстрацiї вiдповiднi данi, що пiдтверджують їх еквiвалентнiсть з референтним препаратом, були вiдсутнi, необхiдно надати данi, отриманi на пiдставi дослiджень вiдповiдно до роздiлу XX цього Порядку. Цi дослiдження мають проводитися пiд час знаходження лiкарського засобу на ринку у рамках програми дослiджень бiоеквiвалентностi генеричного лiкарського засобу до референтного, зазначеної у пiдпунктi 6.4.2 пункту 6.4 цього роздiлу.
Вiд заявника не вимагається надання результатiв дослiджень еквiвалентностi генеричного лiкарського засобу до референтного, отриманих на пiдставi дослiджень вiдповiдно до роздiлу XX цього Порядку, у разi, коли заявлений лiкарський засiб застосовується у медичнiй практицi на територiї України принаймнi протягом 10 рокiв, що пiдтверджується вiдповiдними бiблiографiчними даними щодо його ефективностi, безпеки та якостi.
6.13. Перелiк документiв для проведення експертизи матерiалiв реєстрацiйного досьє на традицiйнi лiкарськi засоби та лiкарськi засоби, що виробляються згiдно iз затвердженими прописами, для державної перереєстрацiї визначено у додатку 8 до цього Порядку.
6.14. Для проведення експертизи матерiалiв реєстрацiйного досьє на АФI або дiючi речовини, якi подаються для державної перереєстрацiї разом iз заявою про державну реєстрацiю (перереєстрацiю) АФI або дiючої речовини, за формою, наведеною у додатку 3 до цього Порядку, додатково подаються:
оновленi методи контролю якостi,
оновленi данi про технологiю виробництва та iнформацiя щодо вiдсутностi змiн у методах контролю якостi та технологiї виробництва АФI або дiючих речовин.
6.15. У разi якщо заявник подає до МОЗ заяву про перереєстрацiю лiкарського засобу менше нiж за 90 календарних днiв до закiнчення строку дiї реєстрацiйного посвiдчення, заява подається вiдповiдно до додатка 1 до цього Порядку, а для традицiйних лiкарських засобiв та лiкарських засобiв, що виробляються згiдно iз затвердженими прописами, - до додатка 2 до цього Порядку. Реєстрацiйнi матерiали подаються згiдно з додатками 5, 8 до цього Порядку (для традицiйних лiкарських засобiв та лiкарських засобiв, що виробляються згiдно iз затвердженими прописами), з урахуванням вимог роздiлiв IX, X, XIII, XIV цього Порядку.
VII. ВИМОГИ ДО МАТЕРIАЛIВ РЕЄСТРАЦIЙНОГО ДОСЬЄ (У ФОРМАТI ЧОТИРЬОХ ЧАСТИН)
1. Вимоги до частини I "Резюме досьє" матерiалiв реєстрацiйного досьє
1.1. Адмiнiстративнi данi (I.A.):
Лiкарський засiб, що є об'єктом заяви про перереєстрацiю, iдентифiкується за назвою лiкарського засобу та за назвою АФI або дiючих речовин разом з лiкарською (фармацевтичною) формою, способом введення, силою дiї та кiнцевою формою випуску готового лiкарського засобу, уключаючи упаковку.
Указуються найменування та мiсцезнаходження заявника разом з найменуванням та мiсцезнаходженням виробникiв i виробничих дiльниць, пов'язаних з рiзними стадiями виробництва (уключаючи виробника готового лiкарського засобу i виробникiв дiючих речовин), та за необхiдностi найменування та мiсцезнаходження iмпортера.
Заявник указує кiлькiсть томiв матерiалiв реєстрацiйного досьє, що супроводжує заяву.
До адмiнiстративних даних належать:
копiї лiцензiї на виробництво (якщо згiдно iз законодавством країни виробника лiцензiя на виробництво iснує лише в електронному виглядi (наприклад, США), має бути надана роздрукiвка iз посиланням на вiдповiдний офiцiйний сайт, засвiдчена пiдписом/печаткою заявника) або iншого дозвiльного документа на виробництво заявленої лiкарської форми у країнi виробника (є необхiдними як для повного, так i неповного виробництва, а також для рiзних процесiв фасування, пакування i маркування);
засвiдчена копiя документа, що пiдтверджує вiдповiднiсть виробництва лiкарського засобу вимогам GMP, виданого Держлiкслужбою вiдповiдно до вимог наказу Мiнiстерства охорони здоров'я України вiд 27 грудня 2012 року N 1130 "Про затвердження Порядку проведення пiдтвердження вiдповiдностi умов виробництва лiкарських засобiв вимогам належної виробничої практики", зареєстрованого у Мiнiстерствi юстицiї України 21 сiчня 2013 року за N 133/22665, або гарантiйний лист заявника щодо надання такого документа протягом строку проведення спецiалiзованої експертизи;
копiя реєстрацiйного посвiдчення або iншого дозвiльного документа (Marketing Authorization, Certificate of a Pharmaceutical Product, Free Sale Certificate тощо), виданого уповноваженим органом країни власника реєстрацiйного посвiдчення (заявника) та/або виробника, або iншої країни, регуляторний орган якої керується високими стандартами до якостi, що вiдповiдають стандартам, рекомендованим ВООЗ, перелiк країн, де даний лiкарський засiб зареєстровано/перереєстровано;
копiя короткої характеристики лiкарського засобу/iнструкцiї для медичного застосування, розробленої та затвердженої вiдповiдно до нормативних вимог країни заявника/виробника, вiдповiдно до встановлених вимог;
перелiк країн, у яких було подано заяви на реєстрацiю.
1.2. Коротка характеристика лiкарського засобу, iнструкцiя для медичного застосування (I.B.):
Копiя короткої характеристики лiкарського засобу/iнструкцiї для медичного застосування, затвердженої вiдповiдно до нормативних вимог країни заявника/виробника або iнших країн, регуляторнi органи яких керуються високими стандартами до якостi, що вiдповiдають стандартам, рекомендованим ВООЗ.
Крiм того, заявник надає проект маркування на упаковцi (первиннiй, вториннiй (за наявностi)), iнструкцiю для медичного застосування лiкарського засобу, складенi вiдповiдно до вимог роздiлiв XVI та XVIII цього Порядку.
Заявник пропонує проект короткої характеристики лiкарського засобу, складеної вiдповiдно до вимог роздiлу XVII цього Порядку, структура якої наведена в додатку 14.
1.3. Звiти незалежних експертiв (I.C.):
Звiти експертiв з документацiї стосовно хiмiчних, фармацевтичних та бiологiчних випробувань лiкарського засобу, а також стосовно документацiї про фармако-токсикологiчнi та клiнiчнi випробування повиннi бути пiдготовленi експертами-фахiвцями, зокрема:
експертом-аналiтиком - вiдомостi про те, чи вiдповiдає лiкарський засiб заявленому складу, iз зазначенням та обґрунтуванням методiв контролю, якi використовуються у виробництвi;
експертом-фармакологом або фахiвцем з аналогiчною експериментальною компетенцiєю - данi щодо токсичностi лiкарського засобу та його фармакологiчних властивостей;
експертом-клiнiцистом - висновки про те, чи змiг вiн переконатися, що ефекти лiкарського засобу в осiб, якi його приймали, збiгаються з вiдомостями, наданими заявником вiдповiдно до встановлених вимог; що лiкарський засiб добре переноситься; рекомендацiї вiдносно дозувань, результати обговорення протипоказань та побiчних реакцiй.
При цьому за необхiдностi експерти мають указати пiдстави для використання бiблiографiчних посилань на наукову лiтературу.
Звiти експертiв повиннi мiстити змiстовну експертизу якостi лiкарського засобу, дослiджень, виконаних на тваринах i за участю людини, а також усi данi, якi мають вiдношення до експертизи. Звiт пишеться таким чином, аби при читаннi можна було скласти чiтке уявлення про властивостi, якiсть, пропонованi специфiкацiї та методи контролю, про безпеку, ефективнiсть, переваги та недолiки лiкарського засобу.
Усi важливi деталi узагальнюються в доповненнях до звiтiв експертiв, за змогою, включаючи данi у формi таблиць та графiкiв. Звiти експертiв та резюме повиннi мiстити чiткi перехреснi посилання на iнформацiю, яка мiститься в основнiй документацiї.
Кожен експертний звiт повинен готуватися квалiфiкованим та досвiдченим спецiалiстом. Вiн повинен бути пiдписаний та датований експертом. До звiту додається коротка iнформацiя про освiту, спецiалiзацiю та професiйний досвiд експерта, а також iнформацiя про професiйнi стосунки мiж заявником та експертом.
2. Вимоги до частини II "Хiмiчна, фармацевтична та бiологiчна документацiя" матерiалiв реєстрацiйного досьє
2.1. Загальнi вимоги:
Усi методики випробувань повиннi вiдповiдати сучасному науковому рiвню, а також перевiренi виробником на достовiрнiсть. При цьому повиннi бути наданi результати дослiджень з перевiрки на достовiрнiсть.
Усi процедури випробувань описуються достатньо точно та докладно, аби можна було вiдтворити їх при проведеннi випробування.
Усi спецiальнi прилади та устаткування описуються досить докладно та, за змогою, супроводжуються схемою.
За потреби наводяться склади лабораторних реактивiв iз зазначенням способу їх приготування.
Якщо методики випробувань описанi в Державнiй фармакопеї України або Європейськiй фармакопеї, то їх опис може бути замiнений докладним посиланням на вiдповiдну фармакопею.
2.2. Склад (II.A.):
Матерiали реєстрацiйного досьє повиннi включати опис якiсних та кiлькiсних характеристик усiх складових лiкарського засобу, а також матерiали щодо фармацевтичної розробки. Зазначенi характеристики та матерiали слiд подавати вiдповiдно до наведених нижче вимог.
2.2.1. Якiснi характеристики:
Пiд якiсними характеристиками усiх складових лiкарського засобу мають на увазi зазначення або опис:
дiючих речовин;
допомiжної(их) речовини(н) незалежно вiд її (їх) походження або використовуваної кiлькостi, включаючи барвники, антиоксиданти, консерванти, ад'юванти, стабiлiзатори, згущувачi, емульгатори, ароматизатори, смаковi добавки тощо;
складових речовин оболонок лiкарських засобiв, призначених для внутрiшнього застосування або введення пацiєнтам в iнший спосiб, - капсули, капсули желатиновi, капсули ректальнi тощо.
На доповнення до цих характеристик має бути надана будь-яка необхiдна iнформацiя про первинну упаковку i, у вiдповiдних випадках, про спосiб її укупорення разом з докладними вiдомостями про пристрої для застосування або введення лiкарського засобу, якi будуть постачатися разом з лiкарським засобом.
У разi перереєстрацiї АФI або дiючої речовини у матерiалах реєстрацiйного досьє, що супроводжують заяву, мають бути наведенi такi вiдомостi:
структурна формула, а також обґрунтованi докази структури, наприклад, на основi синтезу та спектральних даних (якщо такi є);
iнформацiя щодо можливої iзомеризацiї iз зазначенням iзомерiв або можливостi утворення полiморфних модифiкацiй;
фiзико-хiмiчнi та iншi властивостi, уключаючи бiологiчну активнiсть;
iнформацiя щодо домiшок.
Для радiофармацевтичних лiкарських засобiв, до яких уводять радiоактивнi мiтки пiсля постачання виробником, АФI або дiючою речовиною вважається та частина складу лiкарської форми, яка призначена для перенесення або зв'язування радiонуклiда. Наводяться докладнi вiдомостi про джерело радiонуклiда. Крiм того, указуються усi сполуки, якi мають першочергове значення для введення радiоактивної мiтки.
У джерелi АФI або дiючими речовинами вважають як первиннi, так i вториннi радiонуклiди.
2.2.2. Загальноприйнята термiнологiя, яку використовують при описi складових речовин лiкарського засобу, означає таке:
для дiючих речовин, якi наведенi в ДФУ або Європейськiй фармакопеї, має бути вказана основна назва, яка наведена на початку вiдповiдної монографiї, з посиланням на вiдповiдну фармакопею;
для iнших дiючих речовин має бути наведена МНН, рекомендована ВООЗ, яка може супроводжуватися iншою непатентованою назвою або за її вiдсутностi - точною науковою (хiмiчною) назвою; для дiючих речовин, якi не мають МНН або точної наукової назви, мають бути наведенi данi про те, яким чином та з чого вони приготованi (за необхiдностi, ця iнформацiя має бути доповнена вiдповiдними докладними вказiвками);
для барвникiв має бути наведений вiдповiдний код "Е" згiдно з Директивою 78/25/ЕЕС "Про зближення законодавчих положень держав-членiв щодо барвникiв, дозволених для застосування у лiкарських засобах".
2.2.3. Кiлькiснi характеристики:
а) для того аби навести кiлькiснi характеристики дiючих речовин лiкарських засобiв необхiдно, залежно вiд вiдповiдної лiкарської форми, указати масу кожної дiючої речовини або кiлькiсть одиниць бiологiчної дiї у розрахунку на одиницю дози або на одиницю маси, або об'єму.
Для дiючих речовин, якi не можна визначити хiмiчним шляхом, указують одиницi бiологiчної активностi або, якщо такi є, мiжнароднi одиницi бiологiчної активностi, установленi ВООЗ. Якщо мiжнароднi одиницi не встановленi, то одиницi бiологiчної активностi повиннi бути вираженi таким чином, аби надати однозначну iнформацiю про активнiсть дiючих речовин. За змогою має бути вказана бiологiчна активнiсть на одиницю маси.
Додатково вказуються:
для iн'єкцiйних форм: маса та кiлькiсть одиниць бiологiчної активностi для кожної дiючої речовини iз розрахунку на об'єм лiкарського засобу в одиничнiй упаковцi, ураховуючи у вiдповiдних випадках об'єм лiкарського засобу, що використовується пiсля його пiдготовки для безпосереднього застосування;
для лiкарських засобiв у формi крапель: маса або кiлькiсть одиниць бiологiчної активностi для кожної дiючої речовини у кiлькостi крапель, яка вiдповiдає 1 мл або 1 г лiкарського засобу;
для сиропiв, емульсiй, гранульованих та iнших лiкарських форм, призначених для введення у вимiряних кiлькостях: маса або кiлькiсть одиниць бiологiчної активностi для кожної дiючої речовини на вiдмiрену кiлькiсть;
б) якщо дiючi речовини присутнi у виглядi складних сполук або похiдних, то слiд визначити їх у кiлькiсному вираженнi, указавши їх сумарну масу, а за необхiдностi - масу активної частини або частин молекули;
в) кiлькiсний вмiст дiючої речовини, яка є сiллю або гiдратом, завжди виражається у перерахуваннi на масу активної частини (частин) молекули;
г) вимога виражати вмiст дiючих речовин в одиницях маси активних частин, як наведено в пiдпунктi "в" цього пункту, не може бути застосована до радiофармацевтичних лiкарських засобiв. Для радiонуклiдiв радiоактивнiсть виражається в бекерелях на певну дату та, якщо потрiбно, на певний час iз зазначенням часового поясу. Зазначається вид випромiнення.
2.3. Фармацевтична розробка (II.A.4.):
Iнформацiя щодо фармацевтичної розробки лiкарського засобу повинна мiстити обґрунтування вибору складу, складових речовин та первинної упаковки, а також пояснення щодо призначення допомiжних речовин у готовому лiкарському засобi. Обґрунтування має супроводжуватись науковими даними фармацевтичних дослiджень. Необхiдно зазначити надлишки в процесi виробництва з їх обґрунтуванням. Крiм того, мають бути зазначенi характеристики технологiчного процесу (критичнi параметри), якi можуть впливати на вiдтворюванiсть вiд серiї до серiї, функцiональнi характеристики та якiсть лiкарського засобу. До цього роздiлу можуть бути включенi публiкацiї лiтературних джерел.
Повинна бути надана iнформацiя щодо придатностi системи упаковка/укупорка з точки зору вибору матерiалiв, захисту вiд вологи та свiтла, сумiсностi конструкцiйних матерiалiв з лiкарським засобом, уключаючи сорбцiю контейнером речовин або видалення речовин з контейнера та/або безпеку матерiалiв контейнерiв.
Для радiофармацевтичних лiкарських засобiв має бути додатково наведено обґрунтування хiмiчної/радiохiмiчної чистоти та її взаємозв'язок з бiорозподiлом.
2.4. Метод виготовлення (вiдомостi про технологiю виробництва) (II.B.):
Опис способу виробництва лiкарського засобу має надаватися у виглядi вiдомостей про технологiю виробництва. Вiдомостi про технологiю виробництва лiкарського засобу повиннi вiдповiдати наведеним нижче вимогам.
Опис способу виробництва складається таким чином, щоб дати вiдповiдний короткий огляд характеру дiй, що виконуються пiд час виробництва лiкарського засобу, i повинен мiстити iнформацiю про рiзнi стадiї виробництва, що дає змогу оцiнити, чи можуть процеси, здiйснюванi при виробництвi лiкарського засобу, негативно змiнювати його компоненти. У разi безперервного виробництва - докладну iнформацiю про запобiжнi заходи, що застосовуються для забезпечення однорiдностi готової продукцiї.
З цiєю метою вiдомостi про технологiю виробництва лiкарського засобу повиннi мiстити:
а) виробничу формулу (згiдно з вимогами, наведеними в пунктi 2.4.1 цiєї глави);
б) схему стадiй технологiчного процесу (згiдно з вимогами пункту 2.4.2 цiєї глави);
в) опис усiх стадiй технологiчного процесу (згiдно з вимогами пункту 2.4.2 цiєї глави);
г) iнформацiю про експериментальнi дослiдження з валiдацiї виробничого процесу, якщо використовується нестандартний спосiб виробництва або якщо вiн критичний для продукцiї;
ґ) докладний опис процесiв стерилiзацiї та/або процедур забезпечення асептичних умов (для виробництва стерильних лiкарських засобiв).
2.4.1. Виробнича формула (рецептура) повинна щонайменше мiстити таку iнформацiю:
передбачуваний розмiр серiї (у разi змiнюваного та/або альтернативного розмiру серiї має бути надане вiдповiдне обґрунтування) та данi, якi доводять постiйну вiдповiднiсть готової продукцiї усiм специфiкацiям;
назву i кiлькiсть усiх використовуваних у ходi виробництва речовин, включаючи iнгредiєнти, якi видаляють з продукцiї пiд час технологiчного процесу, наприклад розчинники. Мають бути також указанi речовини, якi можуть використовуватися не завжди (наприклад, кислоти i луги для регулювання pH). Для всiх речовин повиннi бути наданi посилання на стандарти їх якостi. Надлишки повиннi бути вказанi в кiлькiсному вираженнi й обґрунтованi в роздiлi реєстрацiйного досьє, що описує фармацевтичну розробку (пункт 2.3 цiєї глави);
для кожного iнгредiєнта повиннi бути вказанi верхня i нижня межi прийнятностi для дiйсної кiлькостi кожної речовини вiд номiнальної кiлькостi у виробничiй рецептурi на серiю. Для дiючих речовин цi межi прийнятностi повиннi бути в iнтервалi вiд 95 % до 105 % вiд номiнальної кiлькостi; для допомiжних речовин межi прийнятностi - в iнтервалi вiд 90 % до 110 % вiд номiнальної кiлькостi є допустимими без додаткового обґрунтування. Можуть допускатися ширшi межi прийнятностi, якi повиннi бути обґрунтованi доказом, що серiї зi складом, подiбним до значень нижнiх i верхнiх запропонованих меж прийнятностi, продовжують вiдповiдати специфiкацiям на готову продукцiю. Якщо кiлькiсть дiючої речовини, яка повинна використовуватися, розраховують з урахуванням дiйсного кiлькiсного вмiсту в серiї цiєї дiючої речовини (факторизацiя), то це має бути зазначено. Якщо використовують iншi речовини для того, щоб загальна маса серiї готової продукцiї дорiвнювала масi, передбаченiй у виробничiй рецептурi на серiю, то це також має бути зазначено.
2.4.2. Виробничий процес:
а) схема стадiй технологiчного процесу повинна мiстити:
зазначення стадiй та операцiй технологiчного процесу (уключаючи операцiї пакування готової продукцiї);
зазначення, де вихiдну сировину i матерiали вводять у процес;
зазначення критичних стадiй (операцiй) технологiчного процесу, а також точки проведення контролю процесу, випробувань промiжної продукцiї або контролю готової продукцiї;
при перереєстрацiї АФI або дiючої речовини повинна бути наведена схема синтезу iз зазначенням молекулярних формул, маси, дiапазонiв виходiв, хiмiчної структури вихiдних iнгредiєнтiв, промiжної продукцiї, реактивiв, самої АФI або дiючої речовини (включаючи стереохiмiю), режимiв роботи та розчинникiв (за наявностi);
б) опис усiх стадiй технологiчного процесу має бути достатнiй для експертизи того, чи можуть цi процеси негативно вплинути на якiсть лiкарського засобу пiд час виробництва. Опис виробничого процесу, уключаючи упаковку, повинен вiдображати послiдовнiсть стадiй i масштаб виробництва. Представлена iнформацiя повинна свiдчити про те, що з високим ступенем вiрогiдностi забезпечується вiдповiднiсть специфiкацiям кожної одиницi кожної серiї готової продукцiї. Для цього:
новi (нестандартнi) процеси або технологiї та операцiї з пакування, якi безпосередньо впливають на якiсть лiкарського засобу, мають бути описанi бiльш детально. Має бути вказане обладнання, щонайменше його вид (наприклад, барабанний змiшувач, гомогенiзатор на лiнiї) i у вiдповiдних випадках робочi параметри. Надана iнформацiя повинна свiдчити про те, що з високою мiрою iмовiрностi забезпечується вiдповiднiсть специфiкацiям кожної одиницi кожної серiї готової продукцiї;
для стадiй (операцiй) процесу необхiдно вказати вiдповiднi параметри, такi як час, температура або pH. Числовi значення взаємопов'язаних параметрiв повиннi бути представленi як очiкуваний дiапазон. Числовi дiапазони для критичних етапiв повиннi бути обґрунтованi. У деяких випадках необхiдно вказувати умови навколишнього середовища (наприклад, низька вогкiсть для шипучого лiкарського засобу);
наявнiсть альтернативних стадiй виробничого процесу (наприклад, два альтернативних способи стерилiзацiї контейнерiв) повинна супроводжуватися пiдтвердженням, що всi процеси, якi пропонуються, постiйно забезпечуватимуть виробництво готової продукцiї вiдповiдно до специфiкацiй;
повиннi бути описанi точки проведення контролю в процесi виробництва i вiдповiднi межi прийнятностi. Так, мають бути наданi критерiї прийнятностi i випробування (з обґрунтуванням, уключаючи експериментальнi данi), що виконуються на критичних етапах виробничого процесу для гарантiї того, що процес є контрольованим. Потрiбно також надати iнформацiю про якiсть i контроль промiжної продукцiї, що видiляється пiд час виробництва;
має бути наведена iнформацiя про те, яким чином забезпечується однорiднiсть готової продукцiї;
в) для наборiв радiофармацевтичних лiкарських засобiв опис способу виготовлення включає як докладний опис виробництва набору, так i детальнi рекомендацiї щодо його остаточної обробки для одержання радiоактивного лiкарського засобу.
Для радiонуклiдiв наводяться пов'язанi з ними ядернi реакцiї.
2.5. Методи контролю вихiдних матерiалiв (II.C.):
2.5.1. Пiд вихiдною сировиною розумiють усi складовi речовини лiкарського засобу та, за необхiдностi, компоненти його первинної упаковки.
У разi якщо АФI або дiюча речовина, яка виготовляється не самим заявником, не описана в ДФУ або Європейськiй фармакопеї або АФI або дiюча речовина описана в ДФУ або Європейськiй фармакопеї, але її отримують у спосiб, при якому зберiгаються домiшки, не зазначенi в монографiї фармакопеї, та монографiя не придатна для належного контролю якостi АФI або дiючої речовини, то заявник має вжити заходiв, аби докладний опис способу виробництва та перевiрка правильностi процесу були наданi безпосередньо виробником АФI або дiючої речовини. Однак у такому разi виробник АФI або дiючої речовини надає заявниковi всi данi, якi можуть стати необхiдними для того, аби заявник мiг вiдповiдати за даний лiкарський засiб. Виробник надає заявниковi документ, у якому вказується, що вiн гарантує однорiднiсть серiй i не вноситиме змiн у виробничий процес або специфiкацiї без повiдомлення про це заявнику.
Матерiали реєстрацiйного досьє, якi супроводжують заяву на затвердження таких змiн, подаються до Центру.
До матерiалiв реєстрацiйного досьє уключаються результати випробувань, пов'язаних з контролем якостi всiх складових речовин, що використовуються, включаючи аналiз серiй, особливо для АФI або дiючих речовин. Документи мають охоплювати положення, наведенi нижче:
а) вихiднi речовини, наведенi у фармакопеях.
Монографiї ДФУ та Європейської фармакопеї застосовуються до всiх дiючих речовин, якi в них зазначенi.
Для усiх складових речовин, що вiдповiдають вимогам ДФУ або Європейської фармакопеї, уважається, що вони вiдповiдають установленим вимогам. У цьому разi опис аналiтичних методiв може бути замiнений докладним посиланням на вiдповiдну фармакопею.
Однак, якщо вихiдну речовину, указану в ДФУ або Європейськiй фармакопеї, одержують у спосiб, при якому залишаються домiшки, якi не контролюються вiдповiдно до монографiй фармакопеї, то слiд зазначити цi домiшки та їх граничнодопустимий вмiст, а також описати методику їх визначення.
Барвники повиннi вiдповiдати вимогам чинного законодавства.
Мають бути вказанi стандартнi тести, якi виконуються для перевiрки кожної серiї вихiдної сировини. Якщо тести вiдрiзняються вiд наведених у фармакопеї, то необхiдно надати докази про те, що вихiднi речовини вiдповiдають вимогам до їх якостi, якi пред'являються цiєю фармакопеєю.
Якщо специфiкацiя, яка мiститься у ДФУ або Європейськiй фармакопеї, не достатня для гарантiї якостi АФI або дiючої речовини, то Центр може вимагати вiд заявника надання бiльш придатних специфiкацiй.
Центр може надати iнформацiю вповноваженим органам, вiдповiдальним за вiдповiдну фармакопею. Заявник повинен надати вповноваженим органам цiєї фармакопеї докладнi вiдомостi про виявленi недолiки та про додатковi специфiкацiї, що застосовуються.
Якщо вихiдна речовина не описана нi в ДФУ, нi в Європейськiй фармакопеї, то припустимим є її вiдповiднiсть монографiї фармакопеї iншої країни; у таких випадках заявник подає копiю монографiї (за необхiдностi її переклад) та, за потреби, документи перевiрки правильностi вказаних у монографiї методик випробувань;
б) вихiднi речовини, якi не наведенi у фармакопеях (ДФУ або Європейськiй фармакопеї).
Якщо складова речовина не наведена у зазначених фармакопеях, то її слiд описати у виглядi специфiкацiї, до якої входить така iнформацiя, а саме:
назва речовини, наводяться усi торговельнi або науковi синонiми;
опис АФI або дiючої речовини у формi, подiбнiй до застосовуваної у ДФУ або Європейськiй фармакопеї; її супроводжують необхiдними пояснювальними даними, особливо щодо молекулярної структури, а також вiдповiдним описом способу синтезу. Якщо АФI або дiючу речовину можна описати лише за допомогою способу виготовлення, то опис повинен бути достатньо докладним для характеристики АФI або дiючої речовини, постiйної як за складом, так i за дiєю;
методи встановлення тотожностi можуть бути описанi як у виглядi повного опису методик, якi використовуються при виготовленнi АФI або дiючої речовини, так i у формi методик для рутинних випробувань;
тести на чистоту описують вiдносно до сумарної кiлькостi очiкуваних домiшок, особливо тих, якi можуть мати шкiдливий вплив, та, за потреби, тих домiшок, якi при можливому сполученнi з iншими АФI або дiючими речовинами, до яких має вiдношення подана заява, здатнi негативно вплинути на стабiльнiсть лiкарського засобу або викривити результати аналiтичних випробувань;
для складних дiючих речовин рослинного або тваринного/людського походження необхiдно розмежувати випадки, коли наявнiсть потенцiйних фармакологiчних ефектiв потребує хiмiчних, фiзичних або бiологiчних методiв контролю основних складових речовин, та випадки, коли дiючi речовини мiстять одну або кiлька груп речовин, якi мають подiбну активнiсть i щодо яких припустиме визначення сумарної кiлькостi;
при використаннi речовин тваринного/людського походження повиннi бути описанi методи, якi забезпечують вiдсутнiсть потенцiйно патогенних агентiв;
для радiонуклiдiв указуються їх походження, тотожнiсть iзотопу, iмовiрнi домiшки, носiй, застосування та специфiчна активнiсть;
зазначаються особливi застережнi заходи, якщо вони необхiднi при зберiганнi вихiдної речовини, та, за необхiдностi, максимальний перiод зберiгання перед проведенням повторних випробувань;
в) фiзико-хiмiчнi властивостi, якi впливають на бiодоступнiсть.
При загальному описi характеристик АФI або дiючих речовин, вiд яких може залежати бiодоступнiсть лiкарського засобу, незалежно вiд того, чи наведенi вони у фармакопеях, указуються такi данi:
кристалiчна форма та коефiцiєнти розчинностi;
розмiр часток (у вiдповiдних випадках - пiсля їх розпилення);
стан сольватацiї;
коефiцiєнт розподiлу масло/вода (за необхiдностi, якщо ця iнформацiя розглядається як суттєва, додатково можуть бути запитанi також значення pK та pH).
Для речовин, що використовуються виключно у формi розчину, необхiдно вказувати данi лише за попередньою вимогою;
г) для радiофармацевтичних лiкарських засобiв до вихiдної сировини вiдносять i речовини, призначенi для опромiнення;
ґ) при перереєстрацiї АФI або дiючої речовини повиннi бути наданi:
перелiк усiх матерiалiв, що використовуються при виготовленнi АФI або дiючої речовини (сировина, вихiднi iнгредiєнти, розчинники, реактиви, каталiзатори тощо) iз зазначенням стадiй (операцiй) технологiчного процесу, на яких вони використовуються;
iнформацiя щодо якостi та контролю цих матерiалiв;
iнформацiя (за необхiдностi), що свiдчить про вiдповiднiсть цих матерiалiв вимогам стандартiв, якi вiдповiдають їх використанню (уключаючи видалення або контроль мiкроорганiзмiв).
2.6. Методи контролю промiжних продуктiв (за необхiдностi) (II.D.):
У матерiалах реєстрацiйного досьє мають мiститися докладнi вiдомостi про контрольнi випробування продукту на промiжних стадiях виробничого процесу з метою забезпечення сталостi технiчних характеристик та технологiчного процесу.
Цi випробування мають бiльше значення для контролю вiдповiдностi лiкарського засобу його складу, коли у виняткових випадках заявник пропонує для випробувань готової продукцiї аналiтичний метод, який не передбачає кiлькiсного визначення усiх дiючих речовин (або усiх допомiжних складових речовин, до яких пред'являються тi самi вимоги, що й до дiючих речовин). Це застосовується i в разi, коли контроль якостi готової продукцiї залежить вiд контрольних випробувань, якi виконуються в процесi виробництва, особливо якщо лiкарський засiб, переважно, характеризують за способом його одержання.
Для лiкарських засобiв тваринного походження заявник повинен продемонструвати, що лiкарський засiб тваринного походження виготовлений в умовах, що зводять до мiнiмуму ризик передачi губчатої енцефалопатiї.
2.7. Методи контролю якостi готового лiкарського засобу (II.E.):
2.7.1. При контролi готового лiкарського засобу вважається, що до його серiї належать усi одиницi даної лiкарської форми, якi були виготовленi з однiєї кiлькостi сировини i пiдлягали однаковiй послiдовностi виробничих операцiй та/або стерилiзацiї, або при безперервному технологiчному процесi всi одиницi готової продукцiї, виготовленi за певний час.
У матерiалах реєстрацiйного досьє повинна мiститися специфiкацiя, згiдно з якою заявник (виробник) здiйснює контроль якостi цього лiкарського засобу. У специфiкацiї повиннi бути зазначенi всi випробування, якi регулярно виконуються для кожної серiї готової продукцiї. Для випробувань, якi виконуються нерегулярно, указується частота їх проведення. Також указуються граничнi значення характеристик показникiв для продукцiї, що випускається.
Матерiали реєстрацiйного досьє (специфiкацiї) повиннi включати докладнi вiдомостi про контрольнi випробування готового лiкарського засобу, якi проводяться на етапi видачi дозволу серiї на випуск. Вони повиннi вiдповiдати вимогам, наведеним нижче:
загальнi характеристики готового лiкарського засобу;
iдентифiкацiя та кiлькiсне визначення дiючої(их) речовини(н);
iдентифiкацiя та кiлькiсне визначення допомiжних речовин;
випробування безпеки;
вимоги до пакувального матерiалу (внутрiшня/зовнiшня упаковка) лiкарського засобу.
Положення монографiй ДФУ або Європейської фармакопеї на лiкарськi форми та радiофармацевтичнi лiкарськi засоби застосовуються до всiх визначених у них лiкарських засобiв.
Якщо використовуються методи випробувань та граничнодопустимi величини, вiдмiннi вiд наведених у монографiях ДФУ або Європейської фармакопеї, то наводиться пiдтвердження того, що готовий лiкарський засiб при випробуваннi вiдповiдно до вимог цих монографiй вiдповiдає фармакопейним вимогам, якi пред'являються до якостi вiдповiдної лiкарської форми.
2.7.2. Загальнi характеристики готового лiкарського засобу:
Випробування готового лiкарського засобу завжди включають i певнi випробування її загальних показникiв якостi. Цi випробування залежно вiд вимог ДФУ або Європейської фармакопеї пов'язанi з контролем середньої маси i максимальних вiдхилень, механiчними, фiзичними i мiкробiологiчними випробуваннями, визначенням органолептичних характеристик, фiзичних характеристик, таких як щiльнiсть, pH, показник заломлення тощо. Заявник у кожному конкретному випадку вказує на норми i граничнодопустимi значення для кожної з цих характеристик.
Докладно описуються умови проведення випробувань, обладнання/прилади, якi використовуються, та норми, якщо вони не наведенi в монографiях ДФУ або Європейської фармакопеї i якщо описанi у фармакопеях методи не застосовуються.
Крiм того, твердi лiкарськi форми, призначенi для приймання внутрiшньо, пiдлягають дослiдженню in vitro для визначення ступеня вивiльнення та розчинення дiючої речовини або речовин; такi дослiдження здiйснюються також у тих випадках, коли введення лiкарського засобу вiдбувається в iнший спосiб, якщо Центр вважатиме це за необхiдне.
2.7.3. Iдентифiкацiя та кiлькiсне визначення дiючої(их) речовини(н):
Iдентифiкацiя та кiлькiсне визначення дiючої(их) речовини(н) проводяться або на усередненiй репрезентативнiй пробi, вiдiбранiй з виробленої серiї, або на певнiй кiлькостi одиниць дозування, якi аналiзуються окремо.
Максимально припустиме вiдхилення вмiсту дiючої речовини в готовому лiкарському засобi на дату його виробництва не повинно перевищувати ±5 %, за винятком вiдповiдним чином обґрунтованих випадкiв.
На основi проведених випробувань стабiльностi виробник повинен запропонувати та обґрунтувати граничнодопустиму кiлькiсть дiючої речовини в готовому лiкарському засобi аж до закiнчення зазначеного термiну зберiгання.
В окремих випадках, якщо лiкарський засiб має особливо складний склад i кiлькiсне визначення дiючих речовин, яких багато або якi присутнi в дуже малих кiлькостях, потребує проведення складних дослiджень, що важко виконати для кожної виробленої серiї, то кiлькiсне визначення однiєї або декiлькох дiючих речовин, якi мiстяться у готовому лiкарському засобi, можна не проводити за умови, що такi кiлькiснi визначення проводять на промiжних стадiях технологiчного процесу. Це положення може не розповсюджуватися на визначення характеристик цих речовин. Така спрощена методика повинна бути доповнена методом кiлькiсної експертизи, яка дає змогу встановити вiдповiднiсть лiкарського засобу, що надiйшов у продаж, його специфiкацiям.
Якщо фiзико-хiмiчнi методи не можуть надати необхiдну iнформацiю про якiсть лiкарського засобу, обов'язково проводиться кiлькiсне визначення бiологiчними методами in vivo та in vitro. При таких дослiдженнях використовуються стандартнi речовини i статистичний аналiз, який дає змогу визначити довiрчi границi вiдповiдного показника. Якщо такi випробування неможливо виконати щодо готового лiкарського засобу, їх проводять на промiжнiй стадiї якомога ближче до закiнчення виробничого процесу.
Якщо вiдповiдно до докладних вiдомостей, наведених у роздiлi IIB, показано, що при виробництвi лiкарського засобу використовують значний надлишок АФI або дiючої речовини, то опис контрольних випробувань готового лiкарського засобу повинен включати хiмiчне, а за необхiдностi й фармако-токсикологiчне, вивчення змiн, яким була пiддана ця АФI або дiюча речовина, а також, можливо, визначення характеристик та/або кiлькiсне визначення продуктiв розпаду.
2.7.4. Iдентифiкацiя та кiлькiсне визначення допомiжних речовин:
Допомiжнi речовини за необхiдностi пiддаються тестам на тотожнiсть.
Пропонованi методики визначення тотожностi барвникiв повиннi дати змогу встановити їх вiдповiднiсть перелiку барвникiв, наведеному у наказi Мiнiстерства охорони здоров'я України вiд 19 червня 2007 року N 339 "Про затвердження Перелiкiв назв допомiжних речовин та барвникiв, що входять до складу лiкарських засобiв" та у додатку до Директиви Ради 78/25/ЄЕС вiд 12 грудня 1977 року "Про зближення правил країн ЄС щодо барвникiв, дозволених для застосування у лiкарських засобах".
Для консервантiв є обов'язковими випробування щодо визначення верхньої та нижньої меж вмiсту. Для iнших допомiжних речовин, здатних негативно впливати на фiзiологiчнi функцiї, обов'язковим є визначення верхньої межi. Якщо допомiжна речовина може вплинути на бiодоступнiсть дiючої речовини, то обов'язковим є визначення верхньої та нижньої меж її вмiсту, за винятком випадкiв, коли необхiдна бiодоступнiсть гарантується iншими вiдповiдними випробуваннями.
2.7.5. Випробування безпеки:
Крiм фармако-токсикологiчних випробувань, якi надаються у матерiалах реєстрацiйного досьє, в аналiтичних документах наводяться докладнi вiдомостi про випробування безпеки, такi як визначення стерильностi, бактерiальних ендотоксинiв, пiрогенностi та мiсцевої переносностi на тваринах, мiкробiологiчної чистоти у тих випадках, коли випробування необхiдно виконувати регулярно для пiдтвердження якостi лiкарського засобу.
Для радiофармацевтичних лiкарських засобiв описуються чистота радiонуклiдiв, радiохiмiчна чистота та специфiчна активнiсть. Вiдхилення вiд значення радiоактивностi, указаного на етикетцi, не повинно перевищувати ±10 %. Для джерел потрiбнi докладнi вiдомостi про випробування первинних та вторинних радiонуклiдiв. Для генераторiв-елюатiв повиннi бути передбаченi випробування первинних радiонуклiдiв та iнших компонентiв системи генератора.
Для наборiв специфiкацiї на готовий лiкарський засiб включають випробування характеристик лiкарського засобу пiсля введення радiоактивної мiтки.
Повинен бути передбачений вiдповiдний контроль радiохiмiчної чистоти та чистоти радiонуклiдiв мiченої сполуки. Необхiдно проводити якiсне та кiлькiсне визначення будь-якої речовини, яка має суттєве значення для введення радiоактивної мiтки.
2.7.6. Вимоги до упаковки:
Повинен бути наведений опис системи упаковка/укупорка, уключаючи зазначення матерiалiв, з яких виготовлено кожний компонент первинної упаковки, а також зазначена специфiкацiя для контролю їх якостi.
Для нефункцiональних компонентiв вторинної упаковки (наприклад, для тих, що не забезпечують додаткового захисту) достатньо наводити лише скорочений опис. Для функцiональних компонентiв вторинної упаковки повинна бути наведена докладна iнформацiя.
Для пристроїв для введення лiкарських засобiв та дозуючих пристроїв необхiдно надати CE-сертифiкат (пiдтвердження вiдповiдностi Директивi 93/42/ЕЕС "Медичнi прилади. Медичне обладнання. Медичнi пристрої"), за наявностi.
2.8. Стабiльнiсть (II.F.):
У матерiалах реєстрацiйного досьє наводиться нижчезазначена iнформацiя. Наводяться опис дослiджень, на пiдставi яких заявник визначив пропонований термiн зберiгання, рекомендованi умови зберiгання та специфiкацiї на дату закiнчення термiну зберiгання.
Якщо в готовому лiкарському засобi можливе утворення продуктiв розпаду, то заявник повинен указати цi продукти розпаду та навести методи визначення їх характеристик та методики випробувань.
Висновки мiстять результати аналiзiв, якi обґрунтовують пропонований термiн зберiгання при рекомендованих умовах зберiгання i специфiкацiї на готовий лiкарський засiб на час закiнчення термiну зберiгання при цих рекомендованих умовах зберiгання.
Указується граничнодопустимий рiвень продуктiв розпаду на дату закiнчення термiну зберiгання.
Якщо можливий ризик взаємодiї лiкарського засобу з первинною упаковкою, особливо в разi iн'єкцiйних лiкарських засобiв або аерозолiв для системного застосування, то надаються результати дослiджень з цього питання.
Для радiофармацевтичних лiкарських засобiв необхiдно наводити iнформацiю щодо стабiльностi джерел радiонуклiдiв, радiонуклiдних наборiв та лiкарських засобiв, якi мiстять радiоактивну мiтку. Слiд задокументовувати стабiльнiсть пiд час використання радiофармацевтичних лiкарських засобiв у багатодозових флаконах.
2.9. Бiодоступнiсть/бiоекiвалентнiсть (II.G.):
Наводяться данi щодо бiодоступностi/бiоеквiвалентностi або даються посилання на вiдповiднi роздiли частини IV матерiалiв реєстрацiйного досьє (за потреби). У цьому разi вiдповiднi роздiли частини IV мають бути включенi до складу матерiалiв реєстрацiйного досьє.
У разi застосування процедури бiовейвер у частинi IV необхiдно надати звiт про проведення дослiджень in vitro. Оцiнка та проведення дослiджень бiоеквiвалентностi або обґрунтування щодо її непроведення мають бути наданi вiдповiдно до вимог роздiлу XX цього Порядку та керiвних настанов.
3. Вимоги до частини III "Фармакологiчна та токсикологiчна документацiя" матерiалiв реєстрацiйного досьє
3.1. Матерiали реєстрацiйного досьє подаються вiдповiдно до наведених нижче вимог.
Вiдповiднiсть дослiджень безпеки лiкарських засобiв забезпечується виконанням положень належної лабораторної практики.
Токсикологiчнi та фармакологiчнi випробування повиннi визначити:
а) потенцiйну токсичнiсть лiкарського засобу та будь-якi небезпечнi або побiчнi токсичнi ефекти, якi можуть спостерiгатися у людини при додержаннi рекомендованих умов застосування лiкарського засобу; ця експертиза повинна бути проведена з урахуванням вiдповiдних патологiчних станiв;
б) фармакологiчнi властивостi лiкарського засобу з урахуванням взаємозв'язку його якiсних та кiлькiсних характеристик з рекомендованими умовами застосування у людини. Усi результати повиннi бути достовiрними та загально прийнятними. При плануваннi експериментальних дослiджень, а також при експертизi одержаних даних використовуються вiдповiднi методи математичної та статистичної обробки результатiв.
Крiм того, необхiдно надати клiнiцистам iнформацiю про потенцiйний терапевтичний ефект лiкарського засобу.
3.2. Якщо лiкарський засiб призначений для мiсцевого застосування, матерiали реєстрацiйного досьє щодо доклiнiчних дослiджень мають мiстити данi щодо дослiджень системної абсорбцiї, з урахуванням можливостi застосування лiкарського засобу на ушкоджених дiлянках шкiри та його всмоктування через iншi вiдповiднi поверхнi. Тiльки в тому випадку, коли доведено, що системна абсорбцiя в даних умовах незначна, можна не дослiджувати системну токсичнiсть у разi використання повторних доз та фетотоксичнiсть, а також вплив на репродуктивну функцiю.
Однак, якщо в ходi дослiджень терапевтичної ефективностi вiдзначається системне всмоктування лiкарського засобу, матерiали реєстрацiйного досьє щодо доклiнiчних дослiджень мають мiстити данi щодо проведених дослiджень токсичностi на тваринах, а, за необхiдностi, фетотоксичностi.
У всiх випадках необхiдно з особливою увагою проводити дослiдження мiсцевої переносностi пiсля застосування повторних доз лiкарського засобу, включаючи гiстологiчнi дослiдження; дослiджувати ймовiрнiсть сенсибiлiзацiї та наявнiсть можливої канцерогенної дiї.
3.3. Для бiологiчних лiкарських засобiв, таких як лiкарськi засоби, одержанi з кровi або плазми кровi людини, заявник повинен обґрунтувати виконану програму випробувань лiкарського засобу.
При плануваннi програми випробувань необхiдно також взяти до уваги таке:
усi випробування, якi потребують повторного введення лiкарського засобу, необхiдно планувати з урахуванням iмовiрної стимуляцiї утворення антитiл та впливу антитiл на органiзм;
слiд розглянути питання про проведення дослiджень репродуктивної функцiї, ембрiональної/фетальної та перинатальної токсичностi, а також можливої мутагенної та канцерогенної дiї. Якщо причиною токсичностi є неактивнi речовини, то можна не проводити дослiдження, якщо доведено, що цi компоненти видаленi з лiкарського засобу.
3.4. Для радiофармацевтичних лiкарських засобiв вважається, що токсичнiсть може бути пов'язана з дозою радiоактивного випромiнення. При дiагностицi це є наслiдком застосування даних лiкарських засобiв, при терапiї - необхiдною властивiстю. При експертизi безпеки та ефективностi радiофармацевтичних лiкарських засобiв ураховуються їх застосування та данi дозиметрiї. Повинен бути задокументований вплив випромiнення на органи та тканини. Визначення дози випромiнення, що поглинається, проводиться вiдповiдно до прийнятої мiжнародної системи залежно вiд особливостей способу введення цих лiкарських засобiв.
3.5. Якщо допомiжна речовина використовується у фармацевтичнiй практицi вперше, необхiдно провести її токсикологiчнi та фармакокiнетичнi дослiдження.
3.6. Якщо iснує iмовiрнiсть значного розпаду лiкарського засобу в процесi його зберiгання, то слiд розглянути питання про проведення токсикологiчного дослiдження продуктiв розпаду.
3.7. Токсичнiсть при однократному введеннi та введеннi повторних доз (III.A.):
3.7.1. Дослiдження токсичностi при однократному введеннi (III.A.1.):
Цi дослiдження призначенi для якiсного та кiлькiсного вивчення гострих токсичних реакцiй, якi можуть виникнути пiсля однократного введення АФI або дiючої речовини або речовин, якi мiстяться в лiкарському засобi, у тих пропорцiях та фiзико-хiмiчному станi, в яких вони присутнi в готовому лiкарському засобi.
Матерiали реєстрацiйного досьє щодо доклiнiчних дослiджень мають мiстити iнформацiю про дослiдження гострої токсичностi, проведенi на двох або бiльше видах ссавцiв, якi належать до вивчених лiнiй, а iнодi й на одному видi тварин за умови обґрунтування причин для прийняття такого рiшення; способiв введення (зазвичай - не менше двох рiзних способiв, один з яких iдентичний або подiбний до того, який рекомендується для введення лiкарського засобу людинi, а iнший забезпечує системну дiю речовини).
Матерiали мають мiстити iнформацiю щодо вивчень рiзних ознак токсичностi, уключаючи мiсцевi реакцiї. Пiд час дослiджень на тваринах має бути отримана максимальна кiлькiсть iнформацiї.
При дослiдженнi токсичностi при однократному введеннi мають бути встановленi ознаки гострої токсичностi та з'ясована причина загибелi тварини, проведена кiлькiсна експертиза приблизної величини летальної дози у тварин вiдповiдних видiв та одержана iнформацiя про залежнiсть ефекту вiд дози, однак при цьому високого ступеня точностi не потрiбно.
Цi дослiдження можуть дати уявлення про ймовiрнi прояви гострого передозування у людини та бути корисними для планування дослiджень токсичностi при введеннi тваринам вiдповiдних видiв повторних доз.
У разi комбiнацiї дiючих речовин матерiали реєстрацiйного досьє мають мiстити iнформацiю щодо проведеного дослiдження з визначення наявностi або вiдсутностi ефекту пiдсилення токсичностi, а також встановлення нових токсичних ефектiв.
3.7.2. Дослiдження токсичностi при повторних введеннях (III.A.2.):
Дослiдження токсичностi при введеннi повторних доз призначенi для виявлення фiзiологiчних та/або патолого-анатомiчних змiн, спричинених повторним введенням дослiджуваної дiючої речовини або комбiнацiї дiючих речовин, та для визначення залежностi цих змiн вiд дози.
Матерiали реєстрацiйного досьє щодо доклiнiчних дослiджень мають мiстити iнформацiю щодо проведених двох видiв (бажано) випробувань: одне короткострокове (протягом 2 - 4 тижнiв), iнше - тривале, тривалiсть якого залежить вiд умов клiнiчного застосування, мета якого - експериментальне визначення нетоксичного дiапазону доз лiкарського засобу, а звичайна тривалiсть - 3 - 6 мiсяцiв. Якщо лiкарський засiб призначений лише для однократного введення пацiєнту, то можлива наявнiсть у матерiалах реєстрацiйного досьє даних лише одного випробування тривалiстю вiд 2 до 4 тижнiв. Якщо тривалiсть дослiдження скорочена або збiльшена дослiдником, таке його рiшення має бути обґрунтоване у матерiалах реєстрацiйного досьє.
Має бути надано обґрунтування доз.
Матерiали реєстрацiйного досьє щодо доклiнiчних дослiджень мають мiстити iнформацiю щодо дослiджень токсичностi при введеннi повторних доз, проведених на двох видах ссавцiв, один з яких не повинен належати до гризунiв.
Вибiр способу введення лiкарського засобу залежить вiд рекомендованого терапевтичного застосування та можливостi системної абсорбцiї лiкарського засобу. Слiд чiтко обумовити метод та частоту введення доз.
Максимальна доза має бути пiдiбрана таким чином, аби можна було виявити шкiдливий вплив лiкарського засобу. Тодi нижчi дози допоможуть визначити переноснiсть лiкарського засобу для тварин.
При проведеннi дослiджень на дрiбних гризунах експеримент та методики контролю мають вiдповiдати масштабам проблеми, що вивчалася, та дати змогу визначити довiрчi межi.
Експертиза матерiалiв доклiнiчних вивчень щодо токсичних ефектiв повинна базуватися на вивченнi поведiнки тварин та їх зросту, гематологiчних та бiохiмiчних дослiдженнях, особливо тих, що мають вiдношення до механiзмiв виведення лiкарського засобу, а також на даних розтину та супутнiх гiстологiчних дослiдженнях.
Якщо лiкарський засiб представлений новою комбiнацiєю вiдомих дiючих речовин, дослiджених вiдповiдно до вимог, матерiали реєстрацiйного досьє щодо доклiнiчних дослiджень можуть мiстити iнформацiю щодо внесення дослiдником необхiдних змiн у методику проведення тривалих дослiджень, обґрунтувавши такi змiни. Виняток становлять випадки, коли при дослiдженнi гострої або пiдгострої токсичностi виявлено новi токсичнi ефекти або їх потенцiювання.
3.8. Репродуктивна функцiя (фертильнiсть та загальна характеристика репродуктивної функцiї) (III.B.):
Якщо внаслiдок проведення iнших випробувань одержано данi, якi вказують на те, що лiкарський засiб може спричиняти шкiдливий вплив на потомство або порушувати чоловiчу або жiночу репродуктивну функцiю, слiд провести вiдповiднi випробування.
3.9. Данi щодо ембрiотоксичностi та тератогенностi (III.C.):
Цi дослiдження спрямованi на виявлення токсичного та особливо тератогенного ефектiв, якi спостерiгаються на стадiї розвитку органiзму пiсля заплiднення, внаслiдок введення дослiджуваного лiкарського засобу самкам у перiод вагiтностi.
Хоча до теперiшнього часу такi випробування мали лише обмежене прогностичне значення щодо прийнятностi даних результатiв для людини, вважається, що вони дають важливу iнформацiю при виявленнi токсичних ефектiв, таких як резорбцiя та iншi аномалiї.
Вiдмова вiд проведення таких випробувань у зв'язку з тим, що лiкарський засiб не призначений для жiнок дiтородного вiку або з якихось iнших причин, повинна бути вiдповiдним чином обґрунтована.
Матерiали реєстрацiйного досьє щодо доклiнiчного вивчення ембрiональної/фетальної токсичностi мають мiстити iнформацiю щодо дослiджень на двох видах ссавцiв, один з яких не повинен належати до гризунiв; у пери- та постнатальний перiоди - як мiнiмум на одному видi тварин. Бажано, аби один з використовуваних видiв тварин вiдповiдав тому виду, на якому проводились дослiдження токсичностi при введеннi повторних доз лiкарського засобу.
Матерiали реєстрацiйного досьє щодо доклiнiчного вивчення мають мiстити iнформацiю щодо особливостей випробувань (кiлькiсть тварин, кiлькiсть лiкарського засобу, що вводиться, вибiр часу введення та критерiї експертизи результатiв) та залежать вiд рiвня сучасних наукових знань на момент подання заяви та ступеня статистичної достовiрностi результатiв.
3.10. Мутагенний потенцiал (III.D.):
Метою вивчення мутагенної активностi є виявлення змiн, якi дослiджувана речовина може спричинити в генетичному матерiалi iндивiдiв або в клiтинах та якi можуть призвести до постiйної та спадкової вiдмiнностi нащадкiв вiд своїх пращурiв. Проведення таких дослiджень обов'язкове при вивченнi будь-якої нової речовини.
Об'єм i характер результатiв, а також критерiї їх експертизи залежать вiд рiвня сучасних наукових знань на момент подання заяви.
3.11. Канцерогенний потенцiал (III.E.):
Проведення випробувань з метою виявлення канцерогенної дiї, як правило, потрiбне:
а) для речовин, якi подiбнi за хiмiчною будовою з вiдомими канцерогенами або канцерогенними сполуками;
б) для речовин, якi при проведеннi тривалих токсикологiчних випробувань спричинили пiдозрiлi змiни;
в) для речовин, у яких пiд час вивчення мутагенної активностi або при проведеннi короткострокових випробувань на канцерогеннiсть були одержанi пiдозрiлi результати.
Проведення таких випробувань може знадобитися також для речовин, якi входять до складу лiкарського засобу, який передбачається регулярно приймати протягом тривалого перiоду життя пацiєнта.
Об'єм i характер результатiв, а також критерiї їх експертизи залежать вiд рiвня сучасних наукових знань на момент подання заяви.
3.12. Фармакодинамiка (III.F.):
Фармакодинамiка вивчає змiни функцiй фiзiологiчних систем, якi спричиняє лiкарський засiб, незалежно вiд того, чи є цi функцiї нормальними або модифiкованими з метою експерименту.
Таке вивчення повинно включати два рiзних пiдходи.
По-перше, необхiдно адекватно описати всi види дiї лiкарського засобу, на пiдставi яких рекомендується його терапевтичне застосування. Результати повиннi бути наданi в кiлькiсному вираженнi (наприклад, за допомогою кривих доза-ефект, час-ефект тощо) та за можливостi зiставленi з даними, одержаними при вивченнi речовини з вiдомою активнiстю. Якщо в заявi стверджується, що терапевтична ефективнiсть дослiджуваної речовини вище, то слiд продемонструвати статистично достовiрну рiзницю мiж ефектами.
По-друге, має бути надана загальна фармакологiчна характеристика речовини та посилання на побiчнi реакцiї. Повиннi бути дослiдженi основнi функцiї фiзiологiчних систем. Чим ближчi дози, що спричиняють побiчнi явища, до доз, якi спричиняють основний терапевтичний ефект, у зв'язку з яким речовину планується застосовувати, тим бiльшою повинна бути глибина такого дослiдження.
Експериментальнi методики (за винятком стандартних методик) повиннi бути описанi так докладно, аби їх можна було вiдтворити. Необхiдно зрозумiло викласти експериментальнi результати та довести їх статистичну достовiрнiсть.
Слiд вивчити всi кiлькiснi змiни реакцiй органiзму, якi виникають при повторному введеннi дiючої речовини, за винятком окремих випадкiв, якi переконливо обґрунтованi.
Вивчення комбiнацiй дiючих речовин повинно ґрунтуватися або на фармакологiчних передумовах, або на характерi терапевтичної дiї лiкарського засобу.
У першому випадку дослiдження з вивчення фармакодинамiки демонструють тi взаємодiї, якi роблять таку комбiнацiю значущою для терапевтичного застосування.
У другому випадку, коли наукове обґрунтування такої комбiнацiї речовин базується на експериментальнiй терапiї, дослiдження встановлюють, чи можливо довести в експериментах на тваринах терапевтичну ефективнiсть такої комбiнацiї речовин. Крiм того, ще й значущiсть будь-якого вiдмiченого побiчного явища.
Нова дiюча речовина комбiнованого лiкарського засобу має бути ретельно вивчена.
3.13. Фармакокiнетика (III.G.):
Фармакокiнетичнi дослiдження мають на увазi вивчення усiх процесiв, якi вiдбуваються з дiючою речовиною в органiзмi, та охоплюють вивчення її всмоктування, розподiл, бiотрансформацiю та виведення.
Дослiдження цих рiзних етапiв може виконуватися як за допомогою фiзичних, хiмiчних або бiологiчних методiв, так i за допомогою вивчення фактичної фармакодинамiчної активностi дiючої речовини.
Iнформацiя щодо розподiлу речовини та її елiмiнацiї (тобто щодо бiотрансформацiї та виведення) потрiбна в тих випадках, коли цi данi необхiднi при визначеннi дози лiкарського засобу для людини. Вона повинна бути одержана i для хiмiотерапевтичних речовин (антибiотикiв тощо), i для речовин, застосування яких залежить вiд їх нефармакодинамiчних ефектiв (наприклад, рiзних дiагностичних засобiв тощо).
Дослiдження фармакокiнетики фармакологiчно дiючих речовин є обов'язковим.
Можна не проводити дослiдження фармакокiнетики нової комбiнацiї вiдомих речовин, кожна з яких вже вивчена вiдповiдно до положень цього Порядку, якщо таке рiшення обґрунтоване результатами випробувань токсичностi та експериментальними терапевтичними дослiдженнями.
3.14. Мiсцева переноснiсть (III.H.):
Дослiдження мiсцевої переносностi проводиться з метою вивчення впливу лiкарського засобу (як дiючих, так i допомiжних речовин) на тканини органiзму в тих дiлянках, якi можуть контактувати з лiкарським засобом унаслiдок його введення при клiнiчному застосуваннi. Дослiдження плануються таким чином, щоб можна було вiдрiзнити будь-який механiчний вплив введення або фiзико-хiмiчний вплив лiкарського засобу вiд токсичного або фармакодинамiчного ефекту.
4. Вимоги до частини IV "Клiнiчна документацiя" матерiалiв реєстрацiйного досьє
4.1. Матерiали реєстрацiйного досьє подаються вiдповiдно до зазначених нижче вимог:
Матерiали реєстрацiйного досьє щодо клiнiчних випробувань мiстять данi щодо науково-дослiдницької роботи, метою якої є дослiдження за участю людини як суб'єкта дослiдження, призначеного для виявлення або пiдтвердження клiнiчних, фармакологiчних та/або iнших фармакодинамiчних ефектiв одного або декiлькох дослiджуваних лiкарських засобiв, та/або виявлення побiчних реакцiй на один або декiлька дослiджуваних лiкарських засобiв, та/або для вивчення усмоктування, розподiлу, метаболiзму та виведення одного або кiлькох лiкарських засобiв з метою пiдтвердження його (їх) безпеки та/або ефективностi.
Експертиза матерiалiв реєстрацiйного досьє щодо клiнiчних випробувань базується на даних клiнiчних випробувань, у тому числi клiнiчних фармакологiчних дослiджень, спланованих з метою визначення ефективностi та безпеки лiкарського засобу за звичайних умов його застосування з урахуванням терапевтичних показань для застосування у людини. Терапевтична користь лiкарського засобу повинна переважати потенцiйний ризик.
Документацiя щодо клiнiчних випробувань повинна дати змогу скласти достатньо обґрунтований та науково достовiрний висновок щодо ефективностi та безпеки лiкарського засобу. До матерiалiв реєстрацiйного досьє мають бути наданi результати всiх клiнiчних випробувань як позитивного, так i негативного характеру.
4.2. Клiнiчний досвiд (IV.B.):
Матерiали реєстрацiйного досьє щодо клiнiчних випробувань мають мiстити iнформацiю щодо планування та проведення усiх фаз клiнiчних випробувань, у тому числi дослiджень з бiодоступностi та бiоеквiвалентностi, а також звiтнiсть, якi повиннi вiдповiдати нормам належної клiнiчної практики.
Усi клiнiчнi випробування, iнформацiя про якi мiститься у матерiалах реєстрацiйного досьє, повиннi бути проведенi вiдповiдно до етичних принципiв, установлених в останнiй редакцiї Гельсiнської декларацiї.
4.3. Надання результатiв (IV.C.):
4.3.1. Матерiали реєстрацiйного досьє мають мiстити докладнi звiти про кожне клiнiчне випробування з детальними вiдомостями, достатнiми для складання об'єктивної думки про нього:
протокол випробування, який мiстить обґрунтування, завдання, методи статистичної обробки та методологiю випробування, умови проведення та контролю випробування, а також особливостi дослiджуваного лiкарського засобу;
сертифiкат(и) аудиту, iнспекцiйної перевiрки (якщо такi є);
список дослiдникiв iз зазначенням їх прiзвищ, мiсць їх проживання, посад, квалiфiкацiї та клiнiчних обов'язкiв, зазначенням країни, у якiй проводилося випробування, а також зiбраної iнформацiї про кожного окремого пацiєнта, у тому числi iндивiдуальних реєстрацiйних форм;
заключний звiт, пiдписаний дослiдником, а при багатоцентрових випробуваннях - усiма дослiдниками або дослiдником-координатором (вiдповiдальним дослiдником).
4.3.2. Матерiали реєстрацiйного досьє щодо клiнiчних випробувань мають мiстити клiнiчнi спостереження, резюмованi для кожного випробування iз зазначенням:
а) кiлькостi хворих, якi отримали лiкування, та їх статi;
б) критерiїв вiдбору до груп та вiкового розподiлу в групах дослiджуваних пацiєнтiв та контрольних групах;
в) кiлькостi пацiєнтiв, передчасно виключених з випробувань, iз зазначенням причин, з яких їх участь у випробуваннi було перервано;
г) при проведеннi контрольованих випробувань за вищеперелiчених умов - перелiку осiб, якi складали контрольну групу:
тих, що не отримували лiкування;
отримували плацебо;
отримували iнший лiкарський засiб з вiдомим терапевтичним ефектом;
тих, що отримували iнше, немедикаментозне лiкування;
ґ) частоти побiчних реакцiй, що спостерiгалися;
д) особливостей щодо пацiєнтiв, якi можуть належати до груп ризику, наприклад людей похилого вiку, дiтей, жiнок у перiод вагiтностi або менструацiї, пацiєнтiв, фiзiологiчний або патологiчний стан яких потребує особливого пiдходу;
е) параметрiв та критерiїв експертизи ефективностi та результатiв з урахуванням цих параметрiв або критерiїв;
є) статистичної експертизи результатiв, якщо цього вимагає дизайн випробування та наявнiсть перемiнних чинникiв.
4.3.3. У матерiалах реєстрацiйного досьє щодо клiнiчних випробувань дослiдник у висновках за експериментальними даними повинен висловити свою думку про безпеку лiкарського засобу в умовах його звичайного застосування; про його переноснiсть, ефективнiсть; указати корисну iнформацiю, яка стосується показань та протипоказань, дозування та середньої тривалостi лiкування; особливi заходи безпеки, яких необхiдно дотримуватись при лiкуваннi; клiнiчнi симптоми передозування. При наданнi результатiв багатоцентрових дослiджень у матерiалах реєстрацiйного досьє мають мiститись висновки вiдповiдального дослiдника вiд iменi всiх центрiв, якi проводили дослiдження, щодо безпеки та ефективностi дослiджуваного лiкарського засобу.
4.3.4. Крiм того, у матерiалах реєстрацiйного досьє щодо клiнiчних випробувань мають мiститись спостереження дослiдника щодо:
а) будь-яких ознак звикання, залежностi або виникнення синдрому вiдмiни лiкарського засобу;
б) будь-яких лiкарських взаємодiй, якi спостерiгались при одночасному введеннi лiкарського засобу з iншими лiкарськими засобами;
в) критерiїв, якi слугували пiдставою для виключення окремих пацiєнтiв з випробування;
г) будь-яких випадкiв смертi, якi були вiдмiченi пiд час проведення випробування або в перiод наступного нагляду.
4.3.5. Докладнi звiти про нову комбiнацiю лiкарських дiючих речовин повиннi бути аналогiчними документацiї, яка необхiдна при вивченнi нових лiкарських засобiв, також у них повиннi бути обґрунтованi безпека та ефективнiсть цiєї комбiнацiї.
4.3.6. Повну або часткову вiдсутнiсть даних клiнiчних випробувань необхiдно обґрунтувати. При одержаннi непередбачених результатiв пiд час випробувань слiд провести додатковi токсикологiчнi та фармакологiчнi випробування та дати їм оцiнку.
Якщо лiкарський засiб призначено для тривалого застосування, необхiдно надати докладнi звiти щодо будь-яких змiн фармакологiчної дiї пiсля призначення повторних доз лiкарського засобу, а також вiдносно встановленої тривалостi курсу приймання лiкарського засобу.
4.4. Клiнiчна фармакологiя (IV.D.):
4.4.1. Фармакодинамiка:
У матерiалах реєстрацiйного досьє має бути наведений наявний взаємозв'язок фармакодинамiчної дiї та ефективностi, у тому числi:
залежнiсть "доза-ефект" та її змiни в часi;
обґрунтування доз та способу введення;
механiзм дiї (якщо це можливо).
Необхiдно описати фармакодинамiчну дiю, яка не має вiдношення до ефективностi.
Сам по собi доказ наявностi фармакодинамiчних ефектiв лiкарського засобу в людини не може бути достатньою пiдставою для того, щоб зробити висновки вiдносно якого-небудь конкретного ймовiрного терапевтичного ефекту.
4.4.2. Фармакокiнетика:
У матерiалах реєстрацiйного досьє має бути наведений опис таких фармакокiнетичних характеристик:
усмоктування (швидкiсть та ступiнь);
розподiл;
метаболiзм;
виведення.
Описуються клiнiчно значущi особливостi, у тому числi значення одержаних даних щодо кiнетики лiкарського засобу для визначення режиму його дозування, особливо щодо пацiєнтiв, якi входять до групи ризику, а також вiдмiнностi фармакокiнетики в людини вiд кiнетики лiкарського засобу, що спостерiгалася в доклiнiчних випробуваннях на тваринах.
4.4.3. Лiкарськi взаємодiї:
Якщо лiкарський засiб при рекомендованому застосуваннi буде вводитися водночас з iншими лiкарськими засобами, до матерiалiв реєстрацiйного досьє надаються докладнi звiти про випробування, проведенi з метою виявлення можливої змiни фармакологiчної дiї при їх сумiсному введеннi.
Якщо виявленi фармакодинамiчнi/фармакокiнетичнi взаємодiї дiючої речовини з iншими лiкарськими засобами або iншими речовинами, наприклад, алкоголем, кофеїном, тютюном або нiкотином, якi можуть вживатися водночас, або якщо такi взаємодiї вiрогiднi, то їх слiд описати, особливо з точки зору клiнiчної значущостi i у зв'язку iз заявленими взаємодiями у текстi короткої характеристики лiкарського засобу.
4.5. Бiодоступнiсть/бiоеквiвалентнiсть (IV.E.):
Експертизу матерiалiв реєстрацiйного досьє щодо бiодоступностi слiд проводити в усiх необхiдних випадках, наприклад, якщо терапевтична доза близька до токсичної або якщо в ходi попереднiх випробувань виявлено вiдхилення, якi можуть бути обумовленi фармакокiнетичними властивостями, наприклад, варiабельнiсть усмоктування.
Крiм того, вивчення бiодоступностi проводиться, якщо необхiдно довести бiоеквiвалентнiсть для лiкарських засобiв.
У разi застосування процедури бiовейвер до матерiалiв реєстрацiйного досьє необхiдно надати звiт про проведення дослiджень in vitro. Оцiнка та проведення дослiджень бiоеквiвалентностi або обґрунтування щодо його непроведення мають бути наданi вiдповiдно до вимог роздiлiв XIX та XX цього Порядку та керiвних настанов.
4.6. Клiнiчна ефективнiсть та безпека (IV.F.):
4.6.1. У матерiалах реєстрацiйного досьє щодо клiнiчних випробувань має мiститись iнформацiя щодо контрольованих клiнiчних випробувань та за можливостi рандомiзованих; будь-яка iнша схема проведення клiнiчних випробувань повинна бути обґрунтована; щодо характеру лiкування в контрольних групах, яке може варiювати вiд випадку до випадку та залежить також вiд етичних норм; тому в окремих випадках порiвнюють ефективнiсть нового лiкарського засобу не з дiєю плацебо, а з терапевтичною ефективнiстю уже вiдомого лiкарського засобу.
За будь-якої можливостi й особливо у тих випадках, коли при проведеннi випробувань дiю лiкарського засобу не можна об'єктивно вимiряти, необхiдно надати iнформацiю щодо вжитих заходiв для уникнення систематичної помилки при проведеннi випробувань, у тому числi використання методiв слiпих дослiджень та рандомiзацiї.
4.6.2. У протоколi клiнiчних випробувань, що мiститься у матерiалах реєстрацiйного досьє, мають бути наведенi ретельно описанi статистичнi методи, якi використовуватимуться, та кiлькiсть пацiєнтiв, критерiї їх включення у випробування (у тому числi розрахунки статистичної потужностi випробувань), рiвень значущостi та опис статистичної одиницi (одиницi вiдбору), а також заходи, ужитi для запобiгання систематичнiй помилцi, особливо методи рандомiзацiї. Iнформацiя про включення у клiнiчне випробування великої кiлькостi дослiджуваних осiб не є адекватною замiною контрольованого випробування, проведеного вiдповiдно до вимог належної клiнiчної практики.
4.6.3. Якщо клiнiчнi звiти з ефективностi та безпеки лiкарського засобу за умови додержання рекомендацiй стосовно його застосування недостатньо обґрунтованi з наукової точки зору, вони не є пiдставою для надання висновку щодо ефективностi та безпеки лiкарського засобу.
4.6.4. Значущiсть даних щодо ефективностi та безпеки лiкарського засобу за умови додержання рекомендацiй щодо його застосування суттєво зросте, якщо вони одержанi декiлькома незалежними компетентними дослiдниками.
4.6.5. Експертний звiт, що мiститься у матерiалах реєстрацiйного досьє, має мiстити iнформацiю з оцiнки безпеки, а також значущостi методiв її експертизи для рiзних випробувань.
4.6.6. Усi побiчнi явища, у тому числi патологiчнi вiдхилення показникiв лабораторних дослiджень, повиннi бути наданi у матерiалах реєстрацiйного досьє окремо, особливо щодо:
загальної кiлькостi випадкiв виникнення побiчних реакцiй;
характеру, тяжкостi та первинної обумовленостi побiчного явища.
4.6.7. Експертиза вiдносної безпеки застосування лiкарського засобу з урахуванням побiчних реакцiй повинна бути проведена щодо:
захворювання, при якому лiкарський засiб призначається;
iнших терапевтичних пiдходiв;
конкретних характеристик у рiзних групах пацiєнтiв;
даних доклiнiчних дослiджень з токсикологiї та фармакологiї.
4.6.8. У матерiалах реєстрацiйного досьє щодо клiнiчних випробувань мають бути наданi рекомендацiї щодо умов, за яких лiкарський засiб повинен застосовуватись для зниження частоти розвитку побiчних реакцiй.
4.7. Матерiали реєстрацiйного досьє, якi супроводжують заяву у виняткових випадках (IV.G.):
Якщо заявник може довести, що для лiкарського засобу неможливо надати повнi данi про ефективнiсть i безпечнiсть за нормальних умов використання, оскiльки:
показання, для яких застосовується лiкарський засiб, зустрiчаються вкрай рiдко; у такий спосiб заявник не може iз цих причин надати достатньо даних або
наявнi науковi данi не дають змоги надати повну iнформацiю, або
збiр такої iнформацiї буде суперечити загальноприйнятим принципам медичної етики,
то реєстрацiйне посвiдчення може видаватися з урахуванням певних зобов'язань.
Цi зобов'язання можуть включати таке:
заявник повинен здiйснити певну програму дослiджень у перiод часу, зазначений МОЗ, результати якої мають бути основою для повторної оцiнки спiввiдношення користь/ризик;
лiкарський засiб, що подається на реєстрацiю, має бути рецептурним та в деяких випадках застосовуватися тiльки пiд медичним наглядом, можливо в лiкарнi, а у випадку радiофармацевтичних препаратiв - квалiфiкованим спецiалiстом;
iнструкцiя для медичного застосування має звертати увагу лiкаря на той факт, що наявний опис даного лiкарського засобу є певною мiрою неповним.
4.8. Пiсляреєстрацiйний досвiд (IV.H.):
Якщо лiкарський засiб уже зареєстрований в iнших країнах, то у матерiалах реєстрацiйного досьє надається iнформацiя про побiчнi реакцiї (якщо можна, з указанням їх частоти) на цей лiкарський засiб та лiкарськi засоби, якi мiстять ту саму дiючу речовину (речовини). Також надаються данi з дослiджень безпеки даного лiкарського засобу, якi проводяться у свiтi.
VIII. СПЕЦIАЛЬНI ВИМОГИ ДО МАТЕРIАЛIВ РЕЄСТРАЦIЙНОГО ДОСЬЄ (У ФОРМАТI ЧОТИРЬОХ ЧАСТИН) ЗА РIЗНИМИ ТИПАМИ ЗАЯВ
1. Спецiальнi вимоги до матерiалiв реєстрацiйного досьє для лiкарських засобiв з добре вивченим медичним застосуванням
Для лiкарських засобiв, дiючу(i) речовину(и) яких добре вивчено в медичному застосуваннi з пiдтвердженою ефективнiстю та прийнятним рiвнем безпеки, застосовуються такi спецiальнi вимоги до матерiалiв реєстрацiйного досьє.
Заявник повинен подавати частину I матерiалiв реєстрацiйного досьє за вимогами, наведеними у главi 1 роздiлу VII цього Порядку, та частину II матерiалiв реєстрацiйного досьє за вимогами, наведеними у главi 2 роздiлу VII цього Порядку.
Для матерiалiв реєстрацiйного досьє частин III та IV у докладнiй науковiй бiблiографiї необхiдно вказувати доклiнiчнi та клiнiчнi характеристики лiкарського засобу.
Для пiдтвердження добре вивченого медичного застосування мають застосовуватися спецiальнi вимоги:
а) фактори, якi необхiдно враховувати при визначеннi добре вивченого медичного застосування компонентiв лiкарських засобiв:
час, протягом якого використовується дiюча речовина;
кiлькiснi аспекти використання дiючої речовини;
ступiнь наукового iнтересу у використаннi дiючої речовини (з посиланням на опублiкованi науковi джерела);
послiдовнiсть наукових оцiнок.
Таким чином, для визначення добре вивченого застосування рiзних дiючих речовин може знадобитися оцiнка за рiзнi перiоди часу. Однак у будь-якому разi перiод часу, необхiдний для визначення добре вивченого медичного застосування дiючої речовини, має бути не меншим за 10 рокiв з дати першого систематичного та документованого використання цiєї дiючої речовини як лiкарського засобу;
б) матерiали реєстрацiйного досьє, наданi заявником, повиннi включати всi аспекти оцiнки безпеки та ефективностi, мiстити або давати посилання на огляд вiдповiдної лiтератури з урахуванням перед- та пiсляреєстрацiйних дослiджень та опублiкованої наукової лiтератури стосовно результатiв епiдемiологiчних дослiджень i особливо порiвняльних епiдемiологiчних дослiджень; всю документацiю, як позитивну, так i негативну. Щодо аспектiв про "добре вивчене медичне застосування" необхiдно уточнити, що "бiблiографiчне посилання" на iншi джерела доказiв (пiсляреєстрацiйнi дослiдження, епiдемiологiчнi дослiдження тощо), а не тiльки наявнi данi, що стосуються методiв контролю та випробувань, можуть бути вагомим доказом безпеки та ефективностi лiкарського засобу за умови, що в матерiалах реєстрацiйного досьє чiтко пояснено та обґрунтовано використання цих джерел iнформацiї;
в) необхiдно обґрунтувати, чому може вважатися доведеним прийнятний рiвень безпеки та/або ефективностi, незважаючи на вiдсутнiсть деяких дослiджень;
г) у доклiнiчних та/або клiнiчних оглядах необхiдно пояснити значимiсть будь-яких поданих даних, що стосуються вже зареєстрованого лiкарського засобу, вiдмiнного вiд того, який пропонується до перереєстрацiї. Необхiдно надати обґрунтування з приводу того, чи можна заявлений лiкарський засiб уважати подiбним до вже зареєстрованого лiкарського засобу, незважаючи на iснуючi розбiжностi;
ґ) пiсляреєстрацiйний досвiд використання iнших лiкарських засобiв, що мiстять тi самi компоненти, є особливо важливим, i заявники пiд час надання матерiалiв реєстрацiйного досьє мають придiлити цьому питанню належну увагу.
2. Спецiальнi вимоги до матерiалiв реєстрацiйного досьє для генеричних лiкарських засобiв
2.1. Матерiали реєстрацiйного досьє на генеричнi лiкарськi засоби повиннi мiстити данi, описанi у частинi I матерiалiв реєстрацiйного досьє за вимогами, наведеними у главi 1 роздiлу VII цього Порядку, та частинi II матерiалiв реєстрацiйного досьє за вимогами, наведеними у главi 2 роздiлу VII цього Порядку, за умови, що заявник має дозвiл власника реєстрацiйного посвiдчення оригiнального лiкарського засобу щодо посилання на частини III та IV матерiалiв його реєстрацiйного досьє.
2.2. У випадку, якщо заявник не має дозволу власника реєстрацiйного посвiдчення оригiнального лiкарського засобу щодо посилання на частини III та IV матерiалiв його реєстрацiйного досьє, вiн надає данi, описанi у частинi I матерiалiв реєстрацiйного досьє за вимогами, наведеними у главi 1 роздiлу VII цього Порядку, та частинi II матерiалiв реєстрацiйного досьє за вимогами, наведеними у главi 2 роздiлу VII цього Порядку, а також данi, що доводять бiодоступнiсть i бiоеквiвалентнiсть з оригiнальним лiкарським засобом, за умови, що вiн не є подiбним бiологiчним лiкарським засобом.
Для цих лiкарських засобiв матерiали реєстрацiйного досьє щодо доклiнiчних/клiнiчних оглядiв/резюме (у разi їх проведення) мають включати такi елементи:
обґрунтування того, що лiкарський засiб є по сутi аналогiчним;
короткий опис домiшок, наявних у серiях дiючої(их) речовини(н), а також у готовому лiкарському засобi, який реєструється (у вiдповiдних випадках про продукти розпаду, що виникають при зберiганнi), разом з оцiнкою цих домiшок;
оцiнка дослiджень бiоеквiвалентностi або обґрунтування того, чому дослiдження не проводилися вiдповiдно до вимог роздiлiв XIX та XX цього Порядку та керiвних настанов;
остання опублiкована лiтература, що є актуальною для АФI або дiючої речовини та даного реєстрацiйного досьє. Для цього можна використовувати анотацiї статей наукових журналiв;
кожне твердження в короткiй характеристицi лiкарського засобу, яке до цього не було вiдомо, або не витiкає, виходячи з властивостей лiкарського засобу та/або його терапевтичної групи, необхiдно розглянути в доклiнiчних/клiнiчних оглядах/резюме та пiдтвердити опублiкованою лiтературою та/або додатковими дослiдженнями. При цьому iнструкцiя для медичного застосування генеричного лiкарського засобу має мiстити iнформацiю, iдентичну до тiєї, яка наведена в iнструкцiї для медичного застосування референтного лiкарського засобу. Будь-якi вiдмiнностi iнструкцiї для медичного застосування генеричного лiкарського засобу мають обґрунтовуватись результатами, отриманими при багатоцентрових клiнiчних дослiдженнях;
якщо до складу генеричного лiкарського засобу входять солi, ефiри або похiднi зареєстрованої АФI або дiючої речовини, вiдмiннi вiд тих, що входять до складу референтного лiкарського засобу, заявник повинен надати додатковi данi з метою доведення еквiвалентностi характеристик безпеки та ефективностi;
якщо дiюча речовина генеричного лiкарського засобу має таку саму терапевтичну дiю, що й оригiнальний зареєстрований лiкарський засiб, але ця дiя пов'язана з iншою сiллю/ефiром/комплексом/похiдним, необхiдно пiдтвердити, що при цьому не змiнюється фармакокiнетика, фармакодинамiка та/або токсичнiсть тiєї частини речовини, яка є вiдповiдальною за данi властивостi, i може змiнити профiль безпеки/ефективностi. Якщо таких доказiв не iснує, то дану речовину можна вважати новою дiючою речовиною;
якщо лiкарський засiб призначено для iншого терапевтичного використання, або його представлено в iншiй лiкарськiй формi, або вiн має застосовуватися iншими шляхами введення, або в iнших дозах, або за iншої позологiї, необхiдно надати результати вiдповiдних токсикологiчних i фармакологiчних дослiджень та/або клiнiчних випробувань.
3. Спецiальнi вимоги до матерiалiв реєстрацiйного досьє для лiкарських засобiв з фiксованою комбiнацiєю
Матерiали реєстрацiйного досьє повиннi стосуватися нових лiкарських засобiв, вироблених з використанням принаймнi двох вiдомих АФI або дiючих речовин, якi ранiше не застосовувалися у такiй комбiнацiї з терапевтичною метою.
Для таких лiкарських засобiв необхiдно надати матерiали повного реєстрацiйного досьє (частина I матерiалiв реєстрацiйного досьє за вимогами, наведеними у главi 1 роздiлу VII цього Порядку, частина II матерiалiв реєстрацiйного досьє за вимогами, наведеними у главi 2 роздiлу VII цього Порядку, частина III матерiалiв реєстрацiйного досьє за вимогами, наведеними у главi 3 роздiлу VII цього Порядку, частина IV матерiалiв реєстрацiйного досьє за вимогами, наведеними у главi 4 роздiлу VII цього Порядку).
За можливостi треба надати iнформацiю щодо дiльниць виробництва, допомiжних речовин та оцiнки безпеки.
4. Спецiальнi вимоги до матерiалiв реєстрацiйного досьє за гiбридною заявою про реєстрацiю лiкарського засобу
У такому випадку заявник має подавати частину I матерiалiв реєстрацiйного досьє за вимогами, наведеними у главi 1 роздiлу VII цього Порядку, частину II матерiалiв реєстрацiйного досьє за вимогами, наведеними у главi 2 роздiлу VII цього Порядку.
Матерiали реєстрацiйного досьє частин III та IV мають мiстити звiти про обмеженi доклiнiчнi дослiдження та/або клiнiчнi випробування, що проводилися заявником (залежно вiд певної ситуацiї щодо вiдмiнностей запропонованого лiкарського засобу та референтного препарату (пункт 6.5 роздiлу VI цього Порядку)), та докладнi бiблiографiчнi данi щодо решти необхiдної iнформацiї для пiдтвердження безпеки та/або ефективностi запропонованого лiкарського засобу. Необхiдно надати обґрунтування з приводу того, чи можна запропонований лiкарський засiб уважати подiбним до вже зареєстрованого лiкарського засобу, незважаючи на наявнi розбiжностi.
5. Спецiальнi вимоги до матерiалiв реєстрацiйного досьє для лiкарських засобiв з продукцiї in bulk
Для готових лiкарських засобiв, якi виготовленi за такими стадiями виробничого процесу, як кiнцеве пакування та/або маркування, застосовуються особливi правила при поданнi матерiалiв реєстрацiйного досьє.
Заявник повинен подавати частину I матерiалiв реєстрацiйного досьє за вимогами, наведеними у главi 1 роздiлу VII цього Порядку.
Для частин II, III та IV фармацевтичнi, доклiнiчнi та клiнiчнi характеристики лiкарського засобу необхiдно вказувати у повному обсязi на пiдставi матерiалiв реєстрацiйного досьє виробника продукцiї in bulk.
Вiдповiднi роздiли частини II також мають мiстити додатковi данi та документацiю заявника (власника реєстрацiйного посвiдчення на такий лiкарський засiб) щодо стадiй виробничого процесу, якi ним виконуються.
Крiм того, у вiдповiдних роздiлах матерiалiв реєстрацiйного досьє необхiдно чiтко вказувати найменування та мiсцезнаходження виробничих потужностей виробника продукцiї in bulk (який вiдповiдає за контроль якостi) та виробничих потужностей виробника, який вiдповiдає за випуск серiї. Iнформацiя щодо виробникiв має бути наведена у текстi маркування.
Необхiдно надати пiдтвердження реєстрацiї заявленого лiкарського засобу у споживчiй упаковцi в країнi виробника продукцiї in bulk.
6. Спецiальнi вимоги до матерiалiв реєстрацiйного досьє для гомеопатичних лiкарських засобiв
Даний роздiл стосується особливих вимог до оформлення частин II та III матерiалiв реєстрацiйного досьє на гомеопатичнi лiкарськi засоби.
Спецiальнi вимоги до частини II матерiалiв реєстрацiйного досьє для гомеопатичних лiкарських засобiв:
а) термiнологiя
Латинська назва гомеопатичної сировини, описаної в матерiалах реєстрацiйного досьє, повинна вiдповiдати латинськiй назвi, зазначенiй в Європейськiй фармакопеї, або в разi вiдсутностi - Нiмецькiй гомеопатичнiй фармакопеї (GHP), Гомеопатичнiй фармакопеї США (HPUS), Британськiй гомеопатичнiй фармакопеї (BHP), Гомеопатичнiй фармакопеї Швабе. У вiдповiдних випадках необхiдно надати традицiйно використовувану назву;
б) контроль вихiдних матерiалiв
Опис та данi щодо вихiдних матерiалiв, тобто щодо всiх використаних матерiалiв, уключаючи сировину та промiжнi матерiали, до кiнцевого розведення у готовому лiкарському засобi повиннi бути пiдкрiпленi додатковими даними про гомеопатичну сировину.
Загальнi вимоги до якостi стосуються всiх вихiдних матерiалiв i сировини, а також промiжних етапiв виробничого процесу до кiнцевого розведення у готовому лiкарському засобi. Якщо можна, потрiбно провести дослiдження за наявностi токсичних компонентiв, а також тодi, коли якiсть неможливо контролювати при кiнцевому розведеннi через високий ступiнь розведення. Необхiдно надати повний опис кожної стадiї виробничого процесу, починаючи вiд вихiдних матерiалiв до кiнцевого розведення у готовому лiкарському засобi.
Якщо застосовуються декiлька розведень, цi стадiї розведення необхiдно проводити вiдповiдно до методiв виробництва гомеопатичних засобiв, якi викладено у вiдповiднiй монографiї Європейської фармакопеї, або за її вiдсутностi в iншiй нацiональнiй фармакопеї;
в) методи контролю готового лiкарського засобу
До готових гомеопатичних лiкарських засобiв застосовуються загальнi вимоги до якостi. Будь-якi винятки вимагають ретельного обґрунтування заявником.
Необхiдно iдентифiкувати та проаналiзувати всi компоненти, якi мають потенцiйну токсичнiсть. Якщо неможливо iдентифiкувати та проаналiзувати всi компоненти, якi можуть виявитися токсичними, наприклад, через їх високий ступiнь розведення в готовому лiкарському засобi, потрiбно обґрунтувати якiсть лiкарського засобу за допомогою повної валiдацiї виробничого процесу та процесу розведення;
г) випробування на стабiльнiсть
Необхiдно пiдтвердити стабiльнiсть готового лiкарського засобу. Данi щодо стабiльностi гомеопатичного лiкарського засобу, як правило, дiйснi як для розчинiв, так i для порошкiв, з яких був вироблений даний лiкарський засiб. Якщо iдентифiкацiя або аналiз дiючої речовини неможливi через високий ступiнь розведення, можуть розглядатися данi щодо стабiльностi лiкарського засобу.
Спецiальнi вимоги до частини III матерiалiв реєстрацiйного досьє для гомеопатичних лiкарських засобiв: необхiдно обґрунтувати вiдсутнiсть будь-якої iнформацiї, наприклад, має надаватися обґрунтування, яким чином може доводитися прийнятний рiвень безпеки, незважаючи на те, що вiдсутнi деякi дослiдження.
7. Спецiальнi вимоги до матерiалiв реєстрацiйного досьє для рослинних лiкарських засобiв
До заяви на перереєстрацiю рослинних лiкарських засобiв додаються матерiали повного реєстрацiйного досьє, до якого повиннi бути включенi такi специфiчнi данi.
Положення частини II, включаючи вiдповiднiсть монографiї Європейської фармакопеї, стосуються готових рослинних лiкарських засобiв. При цьому береться до уваги рiвень наукових знань на момент подання заяви.
Стосовно рослинних лiкарських засобiв розглядаються такi специфiчнi аспекти:
а) рослиннi субстанцiї та рослиннi препарати
Щодо номенклатури рослинної субстанцiї необхiдно вказати ботанiчнi характеристики рослини (рiд, вид, рiзновид та автора), хемотип (у вiдповiдних випадках) частин рослини, iншi назви (синонiми, зазначенi в iнших фармакопеях) та лабораторний код.
Щодо номенклатури рослинного препарату необхiдно вказати ботанiчнi характеристики рослини (рiд, вид, рiзновид та автора), хемотип (у вiдповiдних випадках) частин рослини, визначення препарату, спiввiдношення рослинної субстанцiї до рослинного препарату, розчинник(и), iншi назви (синонiми, зазначенi в iнших фармакопеях) та лабораторний код.
При пiдготовцi документiв щодо структури рослинної субстанцiї та рослинного препарату (у вiдповiдних випадках) необхiдно надати iнформацiю про фiзичну форму, опис компонентiв з вiдомою терапевтичною дiєю або маркерiв (молекулярну формулу, вiдносну молекулярну масу, структурну формулу, уключаючи вiдносну та абсолютну стереохiмiю), а також iнформацiю про iншi компоненти.
При пiдготовцi документiв щодо виробництва рослинної субстанцiї у вiдповiдних випадках необхiдно надати iнформацiю про кожного постачальника сировини, включаючи пiдрядникiв, а також iнформацiю про кожну передбачену виробничу дiльницю або устаткування, задiяне при виробництвi/заготовцi та контролi рослинної субстанцiї.
При пiдготовцi документiв щодо виробництва рослинного препарату у вiдповiдних випадках необхiдно вказати iнформацiю про кожного постачальника сировини, уключаючи пiдрядникiв, а також iнформацiю про кожну передбачену виробничу дiльницю або устаткування, задiяне при виробництвi та контролi рослинного засобу.
Щодо опису виробничого процесу рослинної субстанцiї необхiдно надати iнформацiю, у якiй буде вiдповiдним чином описано виробництво субстанцiї та заготiвлю рослинної сировини, уключаючи географiчне мiсце вирощування та культивування лiкарської рослини, умови її збору, висушування та зберiгання.
Щодо опису виробничого процесу та процедури контролю рослинного препарату необхiдно надати iнформацiю, у якiй буде вiдповiдним чином описане виробництво засобу, включаючи опис обробки сировини, розчинникiв i реагентiв, стадiї очищення та стандартизацiї.
Щодо розробки виробничого процесу необхiдно надати (у вiдповiдних випадках) короткий опис розробки рослинної субстанцiї та рослинного препарату з урахуванням передбачуваного шляху застосування та показань. У вiдповiдних випадках має надаватися обговорення результатiв порiвняння фiтохiмiчної будови рослинної субстанцiї та рослинного препарату, що використовувались на пiдтримку бiблiографiчних даних, та рослинної субстанцiї i рослинного препарату, що є дiючою речовиною готового рослинного лiкарського засобу, на який подано заяву про державну перереєстрацiю.
При описi структури та iнших характеристик рослинної субстанцiї за необхiдностi має надаватися iнформацiя про ботанiчнi, макро- та мiкроскопiчнi, фiтохiмiчнi характеристики та бiологiчну активнiсть.
При описi структури та iнших характеристик рослинного препарату за необхiдностi має надаватися iнформацiя про фiтохiмiчнi i фiзико-хiмiчнi характеристики та бiологiчну активнiсть.
За необхiдностi мають надаватися специфiкацiї для рослинної субстанцiї та рослинного препарату.
У вiдповiдних випадках потрiбно надати опис аналiтичних методiв, що використовувалися при випробуваннi рослинної субстанцiї та рослинного препарату.
При описi валiдацiї аналiтичних методик у вiдповiдних випадках потрiбно надати iнформацiю про валiдацiю методик, включаючи експериментальнi данi для методiв аналiзу, якi використовуються при випробуваннях рослинної субстанцiї та рослинного препарату.
При описi аналiзу серiй у вiдповiдних випадках потрiбно надати опис та результати аналiзу серiй рослинних субстанцiй та рослинних препаратiв у такому виглядi, як монографiї фармакопей.
У вiдповiдних випадках потрiбно надати обґрунтування специфiкацiй рослинних субстанцiй i рослинних препаратiв.
У вiдповiдних випадках потрiбно надати iнформацiю про стандартнi зразки або стандартнi препарати, що використовуються при випробуваннях рослинної субстанцiї та рослинного препарату;
б) готовi рослиннi лiкарськi засоби
Короткий опис розробки складу лiкарського засобу повинен уключати iнформацiю про розробку готового рослинного лiкарського засобу з увагою на передбачуваний спосiб застосування та показання. У вiдповiдних випадках потрiбно надати вiдомостi про результати порiвняння фiтохiмiчної будови лiкарського засобу, який використовується на пiдтримку бiблiографiчних даних, та лiкарського засобу, на який подано заяву.
IX. ВИМОГИ ДО МАТЕРIАЛIВ РЕЄСТРАЦIЙНОГО ДОСЬЄ (У ФОРМАТI ЗАГАЛЬНОГО ТЕХНIЧНОГО ДОКУМЕНТА)
1. Вимоги до матерiалiв реєстрацiйного досьє, наведених у Модулi 1 "Адмiнiстративна iнформацiя"
1.1. Змiст:
Необхiдно надати повний змiст модулiв 1 - 5 реєстрацiйного досьє, яке подається разом iз заявою про державну реєстрацiю лiкарського засобу.
1.2. Форма заяви:
У заявi мають бути зазначенi назва лiкарського засобу, який подається на державну реєстрацiю, назва дiючої(их) речовини(н), лiкарська форма, шлях введення, сила дiї (дозування) та форма випуску готового лiкарського засобу, включаючи упаковку.
Зазначаються найменування/П. I. Б. та мiсцезнаходження/мiсце проживання заявника разом з найменуванням/П. I. Б. виробникiв та мiсцезнаходженням виробничих потужностей, задiяних на рiзних стадiях виробництва (включаючи виробника готового лiкарського засобу та виробника(iв) дiючої(их) речовини(н)), а також за потреби iнформацiю щодо iмпортера.
Заявник має визначити тип заяви та вказати, якi зразки (якщо вони додаються) надано.
До адмiнiстративних даних додають:
копiю лiцензiї на виробництво (якщо згiдно iз законодавством країни виробника лiцензiя на виробництво iснує лише в електронному виглядi (наприклад, США), має бути надана роздрукiвка iз посиланням на вiдповiдний офiцiйний сайт, засвiдчена пiдписом/печаткою заявника) або iншого дозвiльного документа на виробництво заявленої лiкарської форми у країнi виробника (є необхiдними як для повного, так i неповного виробництва, а також для рiзних процесiв фасування, пакування i маркування);
засвiдчену копiю документа, що пiдтверджує вiдповiднiсть виробництва лiкарського засобу вимогам GMP, виданого Держлiкслужбою вiдповiдно до вимог наказу Мiнiстерства охорони здоров'я України вiд 27 грудня 2012 року N 1130 "Про затвердження Порядку проведення пiдтвердження вiдповiдностi умов виробництва лiкарських засобiв вимогам належної виробничої практики", зареєстрованого у Мiнiстерствi юстицiї України 21 сiчня 2013 року за N 133/22665, або гарантiйний лист вiд заявника щодо надання такого документа протягом строку проведення спецiалiзованої експертизи (крiм АФI, дiючих речовин);
перелiк країн, де даний лiкарський засiб зареєстрований/перереєстрований;
копiю короткої характеристики лiкарського засобу/iнструкцiї для медичного застосування, розробленої та затвердженої вiдповiдно до нормативних вимог країни заявника/виробника, вiдповiдно до встановлених вимог;
перелiк країн, у яких було подано заяви на реєстрацiю.
У заявi заявник надає докладну iнформацiю про лiкарський засiб, про пропонованого власника реєстрацiйного посвiдчення та виробника(iв), iнформацiю про статус препарату обмеженого застосування (препарату-сироти), науковi консультацiї та програми розробок у педiатрiї та iншу iнформацiю, передбачену додатком 1.
1.3. Коротка характеристика лiкарського засобу, маркування та iнструкцiя для медичного застосування:
1.3.1. Копiя короткої характеристики лiкарського засобу/iнструкцiї для медичного застосування, затвердженої вiдповiдно до нормативних вимог країни заявника/виробника або iнших країн, регуляторнi органи яких керуються високими стандартами якостi, що вiдповiдають стандартам, рекомендованим ВООЗ.
1.3.2. Маркування:
Заявник має надати пропонований текст маркування для первинної та вторинної упаковок, складений згiдно з вимогами роздiлу XVIII цього Порядку.
1.3.3. Iнструкцiя для медичного застосування:
Заявник має надати пропонований текст iнструкцiї для медичного застосування (на паперовому та електронному носiях), складений згiдно з вимогами роздiлу XVI цього Порядку.
1.3.4. Коротка характеристика лiкарського засобу:
Заявник пропонує проект короткої характеристики лiкарського засобу, складений згiдно з вимогами роздiлу XVII цього Порядку.
1.4. Iнформацiя про незалежних експертiв:
Експерти мають надавати резюме iз зауваженнями, зробленими при розглядi документiв та матерiалiв реєстрацiйного досьє, зокрема до Модулiв 3, 4 i 5 (хiмiчна, фармацевтична та бiологiчна документацiя, доклiнiчна документацiя та клiнiчна документацiя вiдповiдно). Резюме незалежного експерта має висвiтлювати критичнi моменти, пов'язанi з якiстю лiкарського засобу, доклiнiчними дослiдженнями та клiнiчними випробуваннями, i мiстити всi данi, необхiднi для оцiнювання.
Цих вимог необхiдно дотримуватися при складаннi загального резюме з якостi, доклiнiчних дослiджень та клiнiчних випробувань, якi включено до Модуля 2 реєстрацiйного досьє на лiкарський засiб. У Модулi 1 має бути надана iнформацiя, пiдписана незалежними експертами, про їх освiту, спецiалiзацiю та професiйний досвiд. В експертiв має бути вiдповiдна квалiфiкацiя.
1.5. Спецiальнi вимоги до рiзних типiв заяв:
Спецiальнi вимоги до рiзних типiв заяв наведено в роздiлi X цього Порядку.
Додаток до Модуля 1. Оцiнка небезпеки для довкiлля.
Заява про державну реєстрацiю лiкарських засобiв за необхiдностi має мiстити резюме оцiнки ризику, у якому оцiнюють можливi ризики для довкiлля через використання та/або утилiзацiю лiкарського засобу, а також наводять пропозицiї щодо вiдповiдної iнформацiї в маркуваннi. Необхiдно розглянути ризик для довкiлля, пов'язаний з вивiльненням лiкарських засобiв, що мiстять генетично модифiкованi органiзми (далi - ГМО), вiдповiдно до вимог Закону України "Про державну систему бiобезпеки при створеннi, випробуваннi, транспортуваннi та використаннi генетично модифiкованих органiзмiв".
Iнформацiя щодо ризику для довкiлля надається у виглядi додатка до Модуля 1.
Iнформацiя включає:
вступ;
копiю письмового дозволу, виданого уповноваженим органом країни виробника, на навмисне вивiльнення ГМО у навколишнє середовище при використаннi з дослiдницькою метою;
данi, включаючи методи виявлення та iдентифiкацiї, а також унiкальний код ГМО i будь-яку додаткову iнформацiю про ГМО або лiкарський засiб, якi мають значення при оцiнцi ризику для довкiлля;
звiт про оцiнку ризику для довкiлля (далi - ОРД), пiдготовлений на пiдставi наявної iнформацiї;
на основi вищевикладеної iнформацiї та ОРД - висновок стосовно вiдповiдної стратегiї управлiння ризиками щодо ОРД i дослiджуваного лiкарського засобу, план монiторингу в пiсляреєстрацiйний перiод та визначення будь-якої спецiальної iнформацiї, яка повинна надаватися в короткiй характеристицi лiкарського засобу, маркуваннi та iнструкцiї для медичного застосування;
вiдповiднi заходи щодо iнформування населення.
Наведена iнформацiя має бути засвiдчена пiдписом автора iз зазначенням дати, даних щодо освiти, стажування та професiйного досвiду. Необхiдно вказати професiйнi стосунки мiж автором i заявником.
2. Вимоги до матерiалiв реєстрацiйного досьє, наведених у Модулi 2 "Резюме Загального технiчного документа"
У цьому модулi наводять резюме хiмiчної, фармацевтичної та бiологiчної документацiї, доклiнiчних та клiнiчних даних, наданих у Модулях 3, 4 i 5 реєстрацiйного досьє на лiкарський засiб, а також у резюме незалежних експертiв.
Необхiдно визначити та проаналiзувати важливi питання. Слiд передбачити узагальненi фактичнi данi, включаючи матерiали у виглядi таблиць. У цих звiтах передбачають перехреснi посилання на таблицi або на iнформацiю, що мiститься в основнiй документацiї, наданiй в Модулi 3 (Якiсть. Хiмiчна, фармацевтична та бiологiчна iнформацiя про лiкарськi засоби, що мiстять хiмiчнi та/або бiологiчнi дiючi речовини), Модулi 4 (Звiти про доклiнiчнi дослiдження) i Модулi 5 (Звiти про клiнiчнi випробування).
Огляди i резюме повиннi вiдповiдати основним принципам i вимогам, викладеним нижче:
2.1. Загальний змiст:
У Модулi 2 надають змiст наукової документацiї, наведеної у Модулях 2 - 5.
2.2. Вступ:
Має бути надана iнформацiя про фармакологiчну групу, механiзм дiї та запропоноване клiнiчне застосування лiкарського засобу.
2.3. Загальне резюме з якостi:
У загальному резюме з якостi слiд надавати огляд iнформацiї, пов'язаної з хiмiчними, фармацевтичними та бiологiчними даними.
Необхiдно звернути особливу увагу на основнi критичнi параметри та питання, пов'язанi з аспектами якостi, а також надати обґрунтування у тих випадках, коли не дотримано вiдповiдних вимог та настанов. Цей документ повинен охоплювати питання i описувати вiдповiднi данi, якi докладно висвiтлено в Модулi 3.
2.4. Огляд доклiнiчних даних:
Необхiдно наводити узагальнену та критичну оцiнку доклiнiчних дослiджень лiкарського засобу на тваринах/in vitro, а також обговорення та обґрунтування стратегiї випробування i за необхiдностi вiдхилення вiд вiдповiдних вимог.
Необхiдно включити оцiнку домiшок i продуктiв розпаду лiкарського засобу разом з їх потенцiйними фармакологiчними та токсикологiчними ефектами, за винятком лiкарських засобiв бiологiчного походження. Слiд розглянути будь-якi розходження у хiральностi, хiмiчнiй формi та чистотi сполук, якi використовувались у доклiнiчних дослiдженнях i у лiкарському засобi, що буде вироблятися.
Для лiкарських засобiв бiологiчного походження необхiдно оцiнити порiвняннiсть матерiалу, використаного у доклiнiчних дослiдженнях, клiнiчних випробуваннях i лiкарському засобi, який буде реєструватися.
Будь-яка нова допомiжна речовина має бути предметом окремої оцiнки з безпеки.
Необхiдно визначити властивостi лiкарського засобу, доведенi у доклiнiчних дослiдженнях, а також представити значення результатiв з безпеки лiкарського засобу для планованого клiнiчного застосування за участю людини.
2.5. Огляд клiнiчних даних:
Огляд клiнiчних даних має мiстити критичний аналiз клiнiчних даних, якi включено у резюме та Модуль 5. Необхiдно надати спосiб клiнiчної розробки лiкарського засобу, включаючи дизайн випробування, рiшення, прийнятi стосовно дослiдження, а також проведення дослiджень.
Необхiдно надати короткий огляд даних клiнiчних дослiджень, включаючи важливi обмежувальнi фактори, а також оцiнку ризику/користi, яка базується на висновках клiнiчних дослiджень, обґрунтувати запропоновану дозу та показання для застосування, виходячи з отриманих клiнiчних даних щодо ефективностi та безпеки, а також оцiнити, як за допомогою короткої характеристики лiкарського засобу та iнших пiдходiв можна оптимiзувати користь та обмежити ризики.
Необхiдно пояснити всi питання щодо ефективностi та безпеки, якi виникають у процесi розробки.
2.6. Резюме доклiнiчних даних:
Резюме доклiнiчних даних потрiбно надавати на основi фактичних результатiв фармакологiчних, фармакокiнетичних i токсикологiчних дослiджень, проведених на тваринах/in vitro, у текстовому форматi та у виглядi таблиць у такому порядку:
вступ;
резюме фармакологiчних даних у текстовому форматi;
резюме фармакологiчних даних у виглядi таблиць;
резюме фармакокiнетичних даних у текстовому форматi;
резюме фармакокiнетичних даних у виглядi таблиць;
резюме токсикологiчних даних у текстовому форматi;
резюме токсикологiчних даних у виглядi таблиць.
2.7. Резюме клiнiчних даних:
Необхiдно надати докладне iз наведенням фактичних даних резюме клiнiчної iнформацiї з вивчення лiкарського засобу, включеної в Модуль 5. Резюме має включати результати всiх бiофармацевтичних дослiджень, дослiджень з клiнiчної фармакологiї, а також дослiджень з клiнiчної ефективностi та безпеки. Необхiдно надати короткий огляд iндивiдуальних дослiджень.
Клiнiчна iнформацiя у виглядi резюме повинна надаватися в такому порядку:
резюме бiофармацевтичних дослiджень та пов'язаних з ними аналiтичних методiв;
резюме дослiджень з клiнiчної фармакологiї;
резюме з клiнiчної ефективностi;
резюме з клiнiчної безпеки;
короткi огляди iндивiдуальних дослiджень.
3. Вимоги до матерiалiв реєстрацiйного досьє, наведених у Модулi 3: "Якiсть. Хiмiчна, фармацевтична та бiологiчна iнформацiя про лiкарськi засоби, що мiстять хiмiчнi та/або бiологiчнi дiючi речовини"
3.1. Формат та подання.
Загальна схема Модуля 3:
змiст;
основнi данi;
дiюча речовина.
Загальна iнформацiя:
назва;
структура;
загальнi властивостi.
Процес виробництва дiючої(их) речовини(н):
виробник(и);
опис виробничого процесу та контролю у процесi виробництва;
контроль матерiалiв;
контроль критичних стадiй i промiжної продукцiї;
валiдацiя процесу та/або його оцiнка;
розробка виробничого процесу.
Опис характеристик дiючої(их) речовини(н):
доказ структури та iншi характеристики;
домiшки.
Контроль дiючої(их) речовини(н):
специфiкацiя;
аналiтичнi методики;
валiдацiя аналiтичних методик;
аналiзи серiй;
обґрунтування специфiкацiї.
Стандартнi зразки та матерiали.
Система упаковки/укупорки.
Стабiльнiсть:
резюме щодо стабiльностi та висновки;
протокол пiсляреєстрацiйного вивчення стабiльностi та зобов'язання щодо стабiльностi;
данi про стабiльнiсть.
Готовий лiкарський засiб.
Опис i склад лiкарського засобу.
Фармацевтична розробка:
компоненти лiкарського засобу;
дiюча(i) речовина(и);
допомiжнi речовини;
лiкарський засiб;
розробка складу;
надлишки;
фiзико-хiмiчнi та бiологiчнi властивостi;
розробка виробничого процесу;
система упаковки/укупорки;
мiкробiологiчнi характеристики;
сумiснiсть.
Виробництво:
виробник(и);
склад на серiю;
опис виробничого процесу та контролю у процесi виробництва;
контроль критичних стадiй i промiжної продукцiї;
валiдацiя процесу та/або його оцiнка.
Контроль допомiжних речовин:
специфiкацiї;
аналiтичнi методики;
валiдацiя аналiтичних методик;
обґрунтування специфiкацiй;
допомiжнi речовини людського або тваринного походження;
новi допомiжнi речовини.
Контроль готового лiкарського засобу:
специфiкацiя(ї);
аналiтичнi методики;
валiдацiя аналiтичних методик;
аналiзи серiй;
характеристика домiшок;
обґрунтування специфiкацiї(й).
Стандартнi зразки та матерiали.
Система упаковки/укупорки.
Стабiльнiсть:
резюме щодо стабiльностi та висновки;
протокол пiсляреєстрацiйного вивчення стабiльностi та зобов'язання щодо стабiльностi;
данi про стабiльнiсть.
Додатки:
примiщення та обладнання (тiльки для бiологiчних лiкарських засобiв);
оцiнка безпеки щодо стороннiх агентiв;
допомiжнi речовини.
Додаткова iнформацiя:
схема валiдацiї виробничого процесу лiкарського засобу;
пристрiй для введення лiкарського засобу;
сертифiкат(и) вiдповiдностi;
для лiкарських засобiв, якi мiстять або в процесi виробництва яких використовують матерiали тваринного та/або людського походження, сертифiкат вiдповiдностi Європейськiй фармакопеї щодо губчатої енцефалопатiї;
посилання на джерела лiтератури.
3.2. Змiст: основнi принципи та вимоги:
а) хiмiчнi, фармацевтичнi та бiологiчнi данi, якi необхiдно надавати, включають для дiючої(их) речовини(н) i готового лiкарського засобу всю iнформацiю про розробку, виробничий процес, характеристики та властивостi, процедуру та вимоги контролю за якiстю, стабiльнiсть, а також опис складу та оформлення готового лiкарського засобу;
б) необхiдно надати двi основнi складовi iнформацiї про дiючу(i) речовину(и) та готовий лiкарський засiб вiдповiдно;
в) у цьому модулi додатково має бути надана докладна iнформацiя про вихiднi матерiали та сировину, якi використовуються при виробництвi дiючої(их) речовини(н) i допомiжних речовин, що входять до складу готового лiкарського засобу;
г) усi методики та методи випробувань, використанi при виробництвi та контролi дiючої(х) речовини(н) та готового лiкарського засобу, мають бути викладенi чiтко i детально, щоб можна було вiдтворити їх при проведеннi контрольних випробувань, на вимогу вповноваженого органу. Усi методи випробувань повиннi вiдповiдати сучасному науковому рiвню, а також бути валiдованими. Слiд надавати результати дослiджень з валiдацiї. Якщо використовуються методи випробувань, включенi до ДФУ або Європейської фармакопеї, то цей виклад необхiдно замiнити вiдповiдним посиланням на монографiю(ї) та загальний(i) роздiл(и);
ґ) для всiх речовин, препаратiв i лiкарських форм, вказаних у монографiях ДФУ або Європейської фармакопеї, необхiдно робити посилання на цi фармакопеї. Стосовно речовин, якi не вказано в цих фармакопеях, мають надаватися посилання на iншi нацiональнi фармакопеї.
Однак, якщо матерiал, зазначений в ДФУ, або Європейськiй фармакопеї, або в iнших нацiональних фармакопеях, одержують способом, при якому можуть виникати домiшки, що не контролюються за монографiєю фармакопеї, то необхiдно вказати цi домiшки та їх допустимi межi, а також надати методику їхнього визначення. У разi якщо специфiкацiя, включена до монографiї ДФУ, або Європейської фармакопеї, або iншої нацiональної фармакопеї, є недостатньою для забезпечення якостi речовини, може знадобитися докладнiша специфiкацiя. Центр може надати iнформацiю вповноваженим органам, вiдповiдальним за вiдповiдну фармакопею. Заявник повинен надати вповноваженим органам цiєї фармакопеї докладнi вiдомостi про виявленi недолiки та про додатковi специфiкацiї, що застосовуються.
Якщо методи аналiзу включено до ДФУ або Європейської фармакопеї, то немає необхiдностi наводити їх повний виклад, достатньо в кожному роздiлi, в якому планувався виклад цього методу, робити вiдповiдне посилання на монографiю(ї) та загальний(i) роздiл(и);
д) якщо вихiднi матерiали та сировина, дiючi або допомiжнi речовини не описано анi в ДФУ, анi в Європейськiй фармакопеї, анi в iнших нацiональних фармакопеях, то може бути прийнятним посилання на монографiю фармакопеї iншої країни. У таких випадках заявник повинен надати копiю монографiї разом з валiдацiєю аналiтичних методик, описаних у монографiї, а також за необхiдностi переклад;
е) якщо дiючу та/або допомiжну(i) речовину(и) i вихiдний матерiал описано в монографiї Європейської фармакопеї, заявник може надати сертифiкат вiдповiдностi Європейськiй фармакопеї у вiдповiдному пунктi цього модуля. Вважається, що сертифiкати вiдповiдностi монографiї Європейської фармакопеї замiняють суттєвi данi у вiдповiдних роздiлах, зазначених у цьому модулi. Виробник речовини повинен пiдтвердити заявнику письмово, що виробничий процес не змiнювався з часу видачi цього сертифiката вiдповiдностi;
є) для добре вивчених дiючих речовин виробник дiючої речовини або заявник може пiдготувати матерiали з:
докладного опису виробничого процесу,
контролю за якiстю в процесi виробництва та валiдацiї процесу,
у виглядi окремого документа, а виробник може подати їх у виглядi мастер-файла на дiючу речовину (матерiали реєстрацiйного досьє (Drug Master File)).
Однак у такому випадку виробник дiючої речовини має надати заявнику готового лiкарського засобу всi данi, якi можуть знадобитися йому. Виробник повинен надати заявнику письмове пiдтвердження, що вiн гарантує вiдповiднiсть мiж серiями (партiями), а також, що вiн не буде вносити змiни у виробничий процес або специфiкацiї, не поiнформувавши про це заявника. Документи та докладну iнформацiю, що супроводжують заяву на внесення такої змiни, необхiдно подавати в уповноваженi органи; цi документи та данi також надаються заявнику в тих роздiлах, де вони стосуються вiдкритої частини мастер-файла на дiючу речовину;
ж) необхiдно описати особливi запобiжнi заходи щодо передачi губчатої енцефалопатiї тварин (сировина, отримана вiд жуйних тварин): на кожнiй стадiї виробничого процесу заявник повинен продемонструвати вiдповiднiсть використаних матерiалiв, посилаючись, зокрема, на Керiвництво з мiнiмiзацiї ризику передачi збудникiв губчатої енцефалопатiї тварин з лiкарськими засобами. Пiдтвердити вiдповiднiсть можна, посилаючись на вищевказаний документ, або надавши сертифiкат вiдповiдностi конкретнiй монографiї Європейської фармакопеї, або шляхом надання наукових даних для обґрунтування цiєї вiдповiдностi;
з) слiд надати iнформацiю про оцiнку ризику потенцiйного зараження стороннiми агентами (незалежно вiд того, мають вони вiрусне походження чи нi) згiдно з вимогами, викладеними в спецiальних керiвництвах, а також у загальних монографiях та загальних роздiлах ДФУ або Європейської фармакопеї;
и) необхiдно описати будь-якi спецiальнi прилади та обладнання, що може застосовуватися на будь-якiй стадiї виробничого процесу та етапi контролю за лiкарським засобом;
i) для пристроїв з введення лiкарських засобiв необхiдно надати CE-сертифiкат (пiдтвердження вiдповiдностi Директивi 93/42/ЕЕС "Медичнi прилади. Медичне обладнання. Медичнi пристрої") за наявностi.
3.3. Особливу увагу придiляють таким пiдроздiлам Модуля 3:
3.3.1. Дiюча(i) речовина(и) (3.2.S.):
Загальна iнформацiя щодо вихiдних матерiалiв та сировини (3.2.S.1):
а) надається iнформацiя про назву дiючої речовини, включаючи рекомендовану МНН, за наявностi - фармакопейну назву, зазначену в ДФУ, Європейськiй фармакопеї, та хiмiчну назву.
Надається структурна формула, включаючи вiдносну та абсолютну стереохiмiю, молекулярна формула та вiдносна молекулярна маса. Для бiотехнологiчних лiкарських засобiв за потреби необхiдно надати схематичну послiдовнiсть амiнокислот i вiдносну молекулярну масу.
Для бiологiчних лiкарських засобiв необхiдно надати перелiк фiзико-хiмiчних та iнших важливих властивостей дiючої речовини, включаючи її бiологiчну активнiсть;
б) у контекстi цього пункту вихiдними матерiалами вважають всi матерiали, з яких виробляються або екстрагуються дiючi речовини.
Для лiкарських засобiв бiологiчного походження за вихiднi матерiали вважають будь-яку речовину бiологiчного походження, таку як мiкроорганiзми, органи та тканини рослинного або тваринного походження, клiтини або рiдини (включаючи кров або плазму) людського або тваринного походження, бiотехнологiчнi клiтиннi компоненти (рекомбiнантнi або нерекомбiнантнi субстрати клiтини, включаючи первиннi клiтини).
У контекстi цього пункту лiкарським засобом бiологiчного походження вважають усi лiкарськi засоби, дiюча речовина яких є бiологiчною речовиною. Так, бiологiчна речовина - це речовина, що виробляється або екстрагується з бiологiчного джерела, для опису та визначення якостi якої необхiдно надавати комбiнацiю фiзичних, хiмiчних та бiологiчних методiв аналiзу разом iз описом процесу виробництва та контролю за ним. Бiологiчними лiкарськими засобами є iмунологiчнi лiкарськi засоби та лiкарськi засоби, що є похiдними кровi та плазми людини; лiкарськi засоби, що отриманi за допомогою бiотехнологiчних методiв (наприклад, технологiї рекомбiнантної ДНК; контрольованої експресiї генiв, що кодують бiологiчно активнi бiлки прокарiотiв i еукарiотiв, у тому числi трансформованих клiтин тварин; методiв отримання гiбридом i моноклональних антитiл тощо), а також препарати прогресивної терапiї.
Будь-якi iншi речовини, якi використовують для виробництва або екстрагування дiючої(их) речовини(н), але з яких цю дiючу речовину безпосередньо не одержують, а саме: реагенти, живильнi середовища, сироватка зародка ембрiона, добавки та буфери, що застосовуються у препаративнiй хроматографiї тощо, вважають вихiдними матерiалами.
3.3.2. Процес виробництва дiючої(их) речовини(н) (3.2.S.2):
а) заявник зобов'язаний надати опис виробничого процесу дiючої речовини. Для адекватного опису процесу виробництва та контролю за ним потрiбну iнформацiю необхiдно викласти у вiдповiдностi до вимог, встановлених цим Порядком;
б) усi матерiали, необхiднi для виробництва дiючої(их) речовини(н), потрiбно перераховувати iз зазначенням стадiї виробництва, на якiй використовується кожний матерiал. Необхiдно надати iнформацiю про якiсть i контроль цих матерiалiв, а також iнформацiю, яка доводить, що всi матерiали вiдповiдають вимогам щодо їхнього запланованого використання.
Необхiдно перерахувати вихiднi матерiали (сировину), а також навести показники їх якостi та методи контролю.
Необхiдно надати найменування/П. I. Б. та мiсцезнаходження виробничих потужностей та вказати обов'язки кожного виробника, включаючи контрактнi виробництва, а також iнформацiю про кожну iз запланованих виробничих дiльниць або лабораторiй;
в) для бiологiчних лiкарських засобiв встановлено такi додатковi вимоги:
необхiдно надати опис та документальне пiдтвердження походження та iсторiї вихiдних матерiалiв.
Стосовно особливих заходiв щодо запобiгання передачi губчатої енцефалопатiї заявник повинен пiдтвердити, що дiюча речовина вiдповiдає, зокрема, Керiвництву з мiнiмiзацiї ризику передачi збудникiв губчатої енцефалопатiї тварин з лiкарськими засобами.
При використаннi банкiв клiтин надаються докази того, що характеристики клiтин залишилися незмiнними при тiй кiлькостi пасажiв, якi використовуються для виробництва, а також протягом наступного перiоду.
Посiвнi матерiали, банки клiтин, пули сироваток або плазми та iншi матерiали бiологiчного походження, а також за можливостi вихiднi речовини, з яких вони отриманi, випробовуються на наявнiсть стороннiх агентiв.
Якщо присутностi потенцiйно патогенних стороннiх агентiв уникнути неможливо, то вихiднi речовини необхiдно використовувати тiльки в тому випадку, коли при подальшiй обробцi буде забезпечуватися видалення та/або iнактивацiя даних стороннiх агентiв, i це має бути доведено валiдацiєю.
Там, де це можливо, виробництво вакцин повинно ґрунтуватись на системi посiвних партiй та вiдомих банкiв клiтин. При виробництвi бактерiальних та вiрусних вакцин характеристики збудника iнфекцiї повиннi бути продемонстрованi на посiвному матерiалi. Крiм того, по живих вакцинах стабiльнiсть характеристики атенуацiї повинна бути продемонстрована на посiвному матерiалi; якщо докази будуть недостатнiми, характеристики атенуацiї також повиннi бути продемонстрованi на стадiї виробництва.
Для лiкарських засобiв, отриманих з кровi або плазми людини, вiдповiдно до положень, викладених у роздiлi X цього Порядку, необхiдно описати та документально пiдтвердити походження, критерiї та методики вiдбору, транспортування та зберiгання вихiдних матерiалiв.
Необхiдно описати виробничi примiщення та обладнання;
г) у вiдповiдному порядку необхiдно надати iнформацiю про методи контролю та критерiї прийнятностi на кожнiй критичнiй стадiї, iнформацiю про якiсть i контроль промiжних продуктiв, а також про процес валiдацiї та/або його аналiз;
ґ) якщо присутностi потенцiйно патогенних стороннiх агентiв уникнути неможливо, то вихiднi речовини необхiдно використовувати тiльки в тих випадках, коли наступна обробка забезпечує їхнє видалення та/або iнактивацiю, що має доводитись у вiдповiдному модулi, який стосується оцiнки вiрусної безпеки;
д) необхiдно передбачити для дiючої речовини опис i обговорення суттєвих змiн, внесених у виробничий процес пiд час розробки, та/або замiни виробничої дiльницi.
3.3.3. Опис характеристик дiючої(их) речовини(н) (3.2.S.3):
Необхiдно надати данi щодо структури та iнших характеристик дiючої(их) речовини(н).
Слiд пiдтвердити структуру дiючої(их) речовини(н), ґрунтуючись на сучасних фiзико-хiмiчних та/або iмунохiмiчних та/або бiологiчних методах, а також надати iнформацiю про домiшки.
3.3.4. Контроль дiючої(их) речовини(н) (3.2.S.4):
Необхiдно надати докладну iнформацiю про специфiкацiї, якi використовуються для посерiйного контролю дiючої(их) речовини(н), обґрунтування вибору цих специфiкацiй, методiв аналiзу та їх валiдацiї.
Необхiдно надати результати контролю окремих серiй, виготовлених на етапi розробки.
3.3.5. Стандартнi зразки або матерiали (3.2.S.5):
Необхiдно визначити та докладно описати стандартнi матерiали та стандартнi зразки. За можливостi необхiдно застосовувати хiмiчнi стандартнi зразки та бiологiчнi стандартнi матерiали, описанi в ДФУ або Європейськiй фармакопеї.
3.3.6. Система упаковки/укупорки (3.2.S.6):
Необхiдно надати опис контейнера та системи упаковки/укупорки i специфiкацiї її компонентiв. За можливостi необхiдно використовувати пакувальнi засоби, якi вiдповiдають вимогам ДФУ або Європейської фармакопеї.
3.3.7. Стабiльнiсть (3.2.S.7):
а) необхiдно надати короткi вiдомостi про тип проведених дослiджень, використанi протоколи i отриманi пiд час дослiджень результати;
б) необхiдно надати оформленi у вiдповiдному форматi докладнi результати дослiдження стабiльностi, включаючи вiдомостi про аналiтичнi методики, якi використовуються для одержання даних, i валiдацiю цих методик;
в) необхiдно надати протокол про дослiдження стабiльностi в пiсляреєстрацiйний перiод та гарантiї заявника щодо стабiльностi.
3.4. Готовий лiкарський засiб (3.2.P.):
3.4.1. Опис i склад лiкарського засобу (3.2.P.1):
Необхiдно надати опис готового лiкарського засобу i його склад. Iнформацiя повинна охоплювати опис лiкарської форми та складу з перелiком усiх компонентiв готового лiкарського засобу, їх кiлькостей в перерахунку на одиницю дози, функцiї в лiкарському засобi:
дiюча(i) речовина(и);
допомiжна(i) речовина(и) незалежно вiд її (їх) походження або кiлькостi, включаючи барвники, консерванти, модифiкатори, стабiлiзатори, згущувачi, емульгатори, смаковi та ароматичнi речовини тощо;
складовi компоненти лiкарської форми, зовнiшнiх оболонок лiкарських засобiв, що потрапляють в органiзм пацiєнта при застосуваннi внутрiшньо або будь-яким iншим шляхом введення (твердi капсули, м'якi капсули, капсули ректальнi, таблетки, вкритi оболонкою, таблетки, вкритi плiвковою оболонкою тощо);
цi вiдомостi необхiдно доповнити будь-якими суттєвими даними, що стосуються типу контейнера та за потреби способу його укупорювання, разом з докладною iнформацiєю про пристрої, за допомогою яких буде використовуватися або вводитися лiкарський засiб i якi поставлятимуться разом з лiкарським засобом.
Вираз "прийнята термiнологiя", який використовується при описi компонентiв лiкарських засобiв, незалежно вiд застосування iнших положень, означає таке:
для речовин, якi наводяться в ДФУ, або Європейськiй фармакопеї, або ж в iнших нацiональних фармакопеях, - основна назва, наведена у заголовку вiдповiдної монографiї, та посилання на конкретну фармакопею;
для iнших речовин - МНН, рекомендована ВООЗ, або за вiдсутностi такої точна наукова назва; для речовин, що не мають МНН або точної наукової назви;
данi про те, яким чином та iз чого вони виробленi та якi добавки у них вводилися, супроводжуючи за потреби цю iнформацiю вiдповiдними деталями;
для барвникiв - вiдповiдний код "Е", визначений Директивою Ради 78/25/ЄЕС вiд 12 грудня 1977 року "Про зближення правил країн ЄС щодо барвникiв, дозволених для застосування у лiкарських засобах" та/або Директивою Європейського Парламенту та Ради 94/36/ЄС вiд 30 червня 1994 року "Про барвники, що застосовуються у харчових продуктах".
Для того, щоб надати "кiлькiсний склад" дiючої(их) речовини(н) у готових лiкарських засобах, необхiдно залежно вiд розглянутої лiкарської форми вказати масу або кiлькiсть одиниць бiологiчної активностi в розрахунку або на одиницю дози, або на одиницю маси, або на одиницю вмiсту для кожної дiючої речовини.
Якщо дiючi речовини представлено у виглядi сполук або похiдних, то необхiдно надати їх кiлькiсне вираження, указавши їхню загальну масу, а за потреби - масу активної частини молекули.
Для лiкарських засобiв, що мiстять дiючу речовину, яку вперше заявлено у складi лiкарського засобу, кiлькiсну характеристику дiючої речовини, що є сiллю або гiдратом, необхiдно завжди зазначати в перерахунку на масу активної частини молекули. Ця кiлькiсна характеристика має бути однаковою у всiх матерiалах реєстрацiйного досьє незалежно вiд країни подання.
Для АФI або дiючої речовини, якi не можна визначати хiмiчним шляхом, указують одиницi бiологiчної активностi або, якщо такi є, мiжнароднi одиницi бiологiчної активностi, установленi ВООЗ. Якщо мiжнароднi одиницi ВООЗ не встановлено, то одиницi бiологiчної активностi потрiбно виражати таким чином, аби надати точну iнформацiю про активнiсть АФI або дiючої речовини, використовуючи, де необхiдно, одиницi Європейської фармакопеї. За можливостi має бути вказана бiологiчна активнiсть на одиницю маси.
3.4.2. Фармацевтична розробка (3.2.P.2):
У цьому роздiлi надається iнформацiя про дослiдження з розробки, проведенi з метою доведення того, що лiкарська форма, склад, виробничий процес, система упаковки/укупорки контейнера, мiкробiологiчнi властивостi та iнструкцiї для медичного застосування вiдповiдають планованому застосуванню, зазначеному в матерiалах реєстрацiйного досьє заявника.
Дослiдження, описанi в цьому роздiлi, мають вiдрiзнятися вiд посерiйних контрольних випробувань, якi проводяться вiдповiдно до специфiкацiй. Необхiдно визначити та описати критичнi параметри складу та характеристики процесу, якi можуть впливати на вiдтворюванiсть серiй, дiю та якiсть лiкарського засобу. За потреби при наданнi додаткових пiдтверджувальних даних необхiдно посилатися на вiдповiднi пункти Модуля 4 (звiти про доклiнiчнi дослiдження) та Модуля 5 (звiти про клiнiчнi випробування) матерiалiв реєстрацiйного досьє:
а) необхiдно обґрунтувати сумiснiсть дiючої речовини з допомiжними речовинами, а також основнi фiзико-хiмiчнi властивостi дiючої речовини, якi можуть вплинути на дiю готового лiкарського засобу, або сумiснiсть рiзних дiючих речовин одна з одною у випадку лiкарських засобiв у виглядi комбiнацiй;
б) необхiдно обґрунтувати вибiр допомiжних речовин, особливо щодо їх вiдповiдних функцiй i концентрацiй;
в) необхiдно надати опис розробки готового лiкарського засобу, беручи до уваги пропонований шлях введення та спосiб застосування;
г) наявнiсть будь-яких надлишкiв у складi має бути обґрунтованою;
ґ) необхiдно вказати та обґрунтувати будь-якi фiзико-хiмiчнi i бiологiчнi властивостi та будь-якi параметри, що стосуються дiї готового лiкарського засобу;
д) необхiдно надати iнформацiю про вибiр i оптимiзацiю виробничого процесу, а також про розбiжностi мiж виробничим процесом, який використовували при виготовленi серiй, задiяних у фазах клiнiчних випробувань, i планованим серiйним процесом виробництва готового лiкарського засобу;
е) необхiдно обґрунтувати придатнiсть контейнера та системи укупорки, яка використовується для зберiгання, перевезення та застосування готового лiкарського засобу. При цьому може знадобитися опис потенцiйної взаємодiї мiж лiкарським засобом i матерiалом контейнера;
є) як для нестерильних, так i для стерильних лiкарських засобiв необхiдно надати та задокументувати мiкробiологiчнi властивостi лiкарської форми вiдповiдно до вимог ДФУ або Європейської фармакопеї;
ж) з метою пiдтвердження вiдповiдної додаткової iнформацiї, яка мiститься у маркуваннi щодо застосування розчинника(iв) чи дозатора, необхiдно обґрунтувати сумiснiсть готового лiкарського засобу з розчинником(ами), призначеним(и) для розведення перед застосуванням, або дозатором.
3.4.3. Процес виробництва лiкарського засобу (3.2.P.3):
а) опис способу виробництва, зазначеного в заявi про державну реєстрацiю лiкарського засобу (додаток 1 до цього Порядку), викладається таким чином, щоб надати адекватне коротке резюме характеру виконуваних операцiй.
З цiєю метою вiн має включати, як мiнiмум:
опис рiзних стадiй виробництва, включаючи контроль у процесi виробництва та вiдповiднi критерiї прийнятностi для оцiнки того, чи можуть процеси, що використовуються при виробництвi, спричинити будь-якi небажанi змiни компонентiв лiкарської форми;
у випадку безперервного виробничого процесу - докладний опис запобiжних заходiв, необхiдних для забезпечення однорiдностi готового лiкарського засобу;
експериментальнi дослiдження з валiдацiї виробничого процесу при використаннi нестандартних методiв виробництва або у випадку, коли процес критичний для лiкарського засобу;
для стерильних лiкарських засобiв - докладний опис iснуючих процесiв стерилiзацiї та/або асептичних процесiв;
детальну виробничу рецептуру (склад на серiю).
Необхiдно надати найменування/П. I. Б. та мiсцезнаходження виробничих потужностей та вказати обов'язки кожного виробника, включаючи контрактнi виробництва, а також iнформацiю про кожну iз запланованих виробничих дiльниць або лабораторiй;
б) необхiдно навести опис аналiтичних методик для контролю якостi лiкарського засобу, якi можуть застосовуватися на промiжних стадiях технологiчного процесу, з метою забезпечення стандартностi виробничого процесу.
Цi методики є важливими з точки зору перевiрки вiдповiдностi лiкарського засобу виробничiй рецептурi, особливо у тих випадках, коли заявник пропонує аналiтичний метод контролю готового лiкарського засобу, який не включає кiлькiсного визначення усiх дiючих речовин (або всiх допомiжних речовин, якi повиннi вiдповiдати таким самим вимогам, що й дiючi).
Це стосується й випадкiв, коли контроль за якiстю готового лiкарського засобу залежить вiд контрольних випробувань у процесi виробництва, особливо в тих випадках, коли метод виготовлення лiкарського засобу суттєво впливає на його якiсть;
в) необхiдно надати опис, документацiю та результати дослiджень з валiдацiї для критичних стадiй виробництва або критичних методiв кiлькiсного визначення, якi використовуються у виробничому процесi.
3.4.4. Контроль допомiжних речовин (3.2.P.4):
а) слiд надати перелiк усiх вихiдних матерiалiв, що використовуються для виробництва допомiжних речовин, iз зазначенням того, на якому етапi процесу застосовується кожний iз них. Має надаватися iнформацiя про якiсть i контроль цих вихiдних матерiалiв, а також iнформацiя, яка свiдчить про те, що матерiали вiдповiдають стандартам з точки зору їх передбачуваного застосування.
Пропонованi методики визначення тотожностi барвникiв повиннi дати змогу встановити їх вiдповiднiсть перелiку барвникiв, наведеному у наказi Мiнiстерства охорони здоров'я України вiд 19 червня 2007 року N 339 "Про затвердження Перелiкiв назв допомiжних речовин та барвникiв, що входять до складу лiкарських засобiв" та Директивi Європейського Парламенту 78/25/ЄЕС вiд 12 грудня 1977 року "Про зближення правил країн ЄС щодо барвникiв, дозволених для застосування у лiкарських засобах" та/або Директивi Європейського Парламенту та Ради 94/36/ЄС вiд 30 червня 1994 року "Про барвники, що застосовуються у харчових продуктах". Крiм того, барвник має вiдповiдати критерiям чистоти, наведеним у Директивi Європейського Парламенту та Ради 95/45/ЄС вiд 26 липня 1995 року "Про встановлення критерiїв чистоти до барвникiв, що застосовуються у харчових продуктах" зi змiнами;
б) для кожної допомiжної речовини мають бути наданi специфiкацiї та їхнє обґрунтування. Необхiдно описати та належним чином валiдувати аналiтичнi методики, що використовуються для контролю їх якостi;
в) особливу увагу необхiдно придiлити допомiжним речовинам людського або тваринного походження.
З метою дотримання особливих заходiв щодо запобiгання передачi збудникiв губчатої енцефалопатiї тварин заявник також повинен пiдтвердити i для допомiжних речовин, що лiкарський засiб виробляється вiдповiдно до, зокрема, Керiвництва з мiнiмiзацiї ризику передачi збудникiв губчатої енцефалопатiї тварин з лiкарськими засобами.
Вiдповiднiсть вимогам вищезгаданого керiвництва можна пiдтвердити, надавши сертифiкат вiдповiдностi конкретнiй монографiї Європейської фармакопеї щодо збудникiв губчатої енцефалопатiї або науковi данi, якi обґрунтовують цю вiдповiднiсть;
г) новi допомiжнi речовини:
Для допомiжних речовин, що використовуються уперше в лiкарському засобi або застосовуються новим шляхом введення, необхiдно надавати повний опис виробництва, властивостей i контролю, з посиланням на пiдтвердженi доклiнiчнi та клiнiчнi данi щодо безпеки. Ця iнформацiя має бути оформлена так, як зазначено вище для дiючої речовини.
Необхiдно надати документ, що мiстить докладну хiмiчну, фармацевтичну та бiологiчну iнформацiю. Ця iнформацiя має бути оформлена так, як зазначено в Модулi 3 стосовно дiючої речовини.
Iнформацiю про нову допомiжну речовину може бути надано у виглядi окремого документа.
Якщо заявник i виробник нової допомiжної речовини не є однiєю й тiєю самою особою, такий окремий документ має надаватися виробником заявнику для подачi до Центру.
Додаткова iнформацiя про результати дослiдження токсичностi нової допомiжної речовини повинна надаватися в Модулi 4 матерiалiв реєстрацiйного досьє.
Результати клiнiчних дослiджень для нової допомiжної речовини слiд описувати в Модулi 5.
3.4.5. Контроль лiкарського засобу (3.2.P.5):
До серiї готового лiкарського засобу, що пiдлягає контролю, належать усi одиницi лiкарської форми, якi виробляли з однiєї кiлькостi сировини та пiддавали однаковiй послiдовностi технологiчних операцiй та/або стерилiзацiї, або в разi безперервного виробничого процесу усi одиницi готової продукцiї, якi виробляли за певний промiжок часу.
Максимально припустиме вiдхилення вмiсту дiючої речовини в готовому лiкарському засобi на дату його виробництва не повинно перевищувати ±5 %, за винятком вiдповiдним чином обґрунтованих випадкiв.
Необхiдно надати докладну iнформацiю про специфiкацiї (при випуску й протягом термiну придатностi) з обґрунтуванням їхнього вибору, методiв аналiзу та валiдацiї.
3.4.6. Стандартнi зразки та матерiали (3.2.P.6):
Необхiдно визначити та докладно описати стандартнi матерiали та стандартнi зразки, якi використовуються пiд час контролю готового лiкарського засобу, якщо про них не вказано в роздiлi, що стосується дiючої речовини.
3.4.7. Система упаковки/укупорки (3.2.P.7):
Необхiдно надати опис контейнера та укупорювальної системи, включаючи матерiали, з яких вироблено кожний компонент первинної упаковки, а також їх специфiкацiї. Специфiкацiї повиннi включати опис i методи контролю. За потреби має надаватись iнформацiя про нефармакопейнi методи (включаючи валiдацiю).
Для нефункцiональних зовнiшнiх пакувальних матерiалiв надається тiльки короткий опис. Для функцiональних компонентiв вторинної упаковки надається додаткова iнформацiя.
Для пристроїв для введення лiкарських засобiв необхiдно надати CE-сертифiкат (пiдтвердження вiдповiдностi Директивi 93/42/ЕЕС "Медичнi прилади. Медичне обладнання. Медичнi пристрої") за наявностi.
3.4.8. Стабiльнiсть (3.2.P.8):
а) необхiдно надати короткi резюме про види проведених дослiджень, використанi протоколи i отриманi пiд час дослiджень результати;
б) необхiдно надати оформленi у вiдповiдному форматi докладнi результати дослiдження стабiльностi, включаючи вiдомостi про аналiтичнi методики, якi використовуються для одержання даних, i валiдацiю цих методик.
Для вакцин потрiбно надати iнформацiю про кумулятивну стабiльнiсть за необхiдностi;
в) необхiдно надати протокол з вивчення стабiльностi у пiсляреєстрацiйний перiод i гарантiї заявника щодо стабiльностi.
4. Вимоги до матерiалiв реєстрацiйного досьє, наведених у Модулi 4 "Звiти про доклiнiчне дослiдження"
4.1. Загальний план Модуля 4:
змiст;
звiти про дослiдження.
Фармакологiя:
первинна фармакодинамiка;
вторинна фармакодинамiка;
фармакологiя безпеки;
фармакодинамiчнi взаємодiї.
Фармакокiнетика:
аналiтичнi методики та звiти щодо їх валiдацiї;
всмоктування;
розподiл;
метаболiзм;
виведення;
фармакокiнетичнi взаємодiї (доклiнiчнi);
iншi фармакокiнетичнi дослiдження.
Токсикологiя:
токсичнiсть при одноразовому введеннi;
токсичнiсть при повторних введеннях;
генотоксичнiсть;
канцерогеннiсть;
репродуктивна токсичнiсть та токсичний вплив на розвиток потомства;
мiсцева переносимiсть.
Додатковi дослiдження токсичностi.
Посилання на джерела лiтератури.
4.2. Звiти про дослiдження.
Особливу увагу необхiдно звернути на таке:
у матерiалах реєстрацiйного досьє щодо фармакологiчних та токсикологiчних випробувань повиннi визначати:
а) потенцiйну токсичнiсть лiкарського засобу та будь-якi небезпечнi або небажанi токсичнi реакцiї, що можуть спостерiгатися за запропонованих умов його застосування людиною; має бути наведена їх оцiнка з урахуванням вiдповiдних патологiчних станiв;
б) фармакологiчнi властивостi лiкарського засобу за якiсними i кiлькiсними показниками стосовно запропонованого клiнiчного застосування. Усi результати повиннi бути достовiрними i мати загальне застосування. При плануваннi експериментальних дослiджень i оцiнцi отриманих даних необхiдно використовувати методи математичної та статистичної обробки результатiв.
Крiм того, у матерiалах реєстрацiйного досьє необхiдно надати iнформацiю про терапевтичний i токсикологiчний потенцiал лiкарського засобу.
У матерiалах реєстрацiйного досьє щодо програми дослiдження необхiдно враховувати, що:
усi дослiдження, що вимагають повторного введення лiкарського засобу, мають плануватися з огляду на ймовiрну стимуляцiю утворення антитiл i впливу антитiл на органiзм;
також необхiдно розглянути питання доцiльностi проведення дослiджень репродуктивної функцiї, ембрiональної/фетальної i перинатальної токсичностi i можливої мутагенної та канцерогенної дiї. Якщо причиною токсичностi є не дiюча речовина, а iншi речовини, то можна не проводити дослiдження, за умови, коли iснує достовiрне пiдтвердження вилучення цих компонентiв з лiкарського засобу.
Якщо допомiжна речовина використовується у фармацевтичнiй практицi вперше, необхiдно провести її токсикологiчнi та фармакокiнетичнi дослiдження.
Якщо iснує ймовiрнiсть значного розпаду лiкарського засобу пiд час його зберiгання, необхiдно розглянути питання про проведення токсикологiчного дослiдження продуктiв розпаду.
4.2.1. Фармакологiя:
У матерiалах реєстрацiйного досьє щодо фармакологiчних дослiджень необхiдно висвiтлити два рiзних пiдходи:
по-перше, фармакодинамiчна активнiсть лiкарського засобу, що пропонується до терапевтичного застосування, має бути вiдповiдним чином дослiдженою i описаною. По змозi повиннi використовуватися визнанi й валiдованi методики дослiдження як in vivo, так i in vitro. Опис нових експериментальних методик повинен бути достатньо докладним, щоб забезпечити їхнє вiдтворення. Результати потрiбно надавати за кiлькiсними показниками, наприклад, кривими доза-ефект та/або час-ефект тощо. Результати повиннi зiставлятися з даними, що характеризують речовину або речовини з аналогiчною терапевтичною дiєю. Вiдсутнiсть порiвняльних дослiджень має бути обґрунтованою;
по-друге, данi про основнi фармакологiчнi властивостi дiючої речовини, iз зазначенням її побiчної дiї на основнi функцiї фiзiологiчних систем органiзму. Якщо дози лiкарського засобу, що викликають негативнi побiчнi реакцiї, є близькими до доз, рекомендованих для медичного застосування, цi дослiдження мають бути поглибленими.
Якщо експериментальнi методи не є стандартними, вони мають бути достатньо детально описаними, аби мати можливiсть їх вiдтворення i пiдтвердження їх достовiрностi. Результати експерименту мають бути чiтко викладеними i доведено їх статистичну достовiрнiсть. Будь-якi кiлькiснi змiни реакцiй, що виникли у вiдповiдь на повторне введення дiючої речовини, мають бути дослiдженими.
Матерiали реєстрацiйного досьє щодо вивчень фiксованих комбiнацiй дiючих речовин стосовно їх фармакодинамiчної взаємодiї повиннi ґрунтуватися або на фармакологiчних передумовах, або на показаннях для їх застосування. У першому випадку фармакодинамiчне дослiдження повинне пiдтвердити взаємодiї, що роблять таку комбiнацiю значимою для терапевтичного застосування. У другому випадку, коли наукове обґрунтування такої комбiнацiї речовин базується на експериментальнiй терапiї, дослiдження має встановити можливiсть пiдтвердження на тваринах дiї, що очiкується вiд такої комбiнацiї речовин, i, принаймнi, значимiсть будь-яких виявлених побiчних реакцiй.
4.2.2. Фармакокiнетика:
Матерiали реєстрацiйного досьє щодо фармакокiнетичних дослiджень включають аналiз всiх процесiв, що вiдбуваються з дiючою речовиною i її метаболiтами в органiзмi, та охоплюють вивчення всмоктування, розподiлу, бiотрансформацiї та виведення цих речовин.
Дослiдження кожного з цих етапiв може виконуватися як за допомогою фiзичних, хiмiчних або бiологiчних методiв, так i шляхом вивчення фактичної фармакодинамiчної активностi самої дiючої речовини.
Iнформацiя про розподiл i виведення з органiзму є необхiдною у всiх випадках, коли такi данi є обов'язковими для визначення доз для людини, та стосовно хiмiотерапевтичних лiкарських засобiв (антибiотикiв тощо) i речовин, використання яких залежить вiд їх нефармакодинамiчних ефектiв (наприклад, численнi дiагностичнi засоби тощо).
Можна також провести дослiдження in vitro, перевагою яких є використання тест-систем людського походження та порiвняння з тест-системами тваринного походження (тобто зв'язування з бiлками, метаболiзм, взаємодiя мiж лiкарськими засобами).
У матерiалах реєстрацiйного досьє необхiдно обов'язково наводити iнформацiю щодо фармакокiнетичних дослiджень фармакологiчно активних речовин. При використаннi нових фiксованих комбiнацiй вiдомих дiючих речовин, що вже були дослiдженими, вiдповiдно до положень цього Порядку iнформацiя щодо фармакокiнетичних дослiджень може не наводитись, якщо таке рiшення обґрунтовано результатами дослiдження токсичностi та експериментальних терапевтичних випробувань.
Дизайн фармакокiнетичних дослiджень має забезпечувати порiвняння даних для тварин i людини та екстраполяцiю на людину результатiв, отриманих для тварин.
4.2.3. Токсикологiя:
а) токсичнiсть при одноразовому введеннi
Матерiали реєстрацiйного досьє щодо дослiдження токсичностi при одноразовому введеннi включають якiсний i кiлькiсний аналiз токсичних проявiв, що можуть виникнути внаслiдок одноразового введення дiючої речовини або речовин, якi мiстяться в лiкарському засобi у таких пропорцiях i фiзико-хiмiчному станi, як i в готовому лiкарському засобi;
б) токсичнiсть при повторних (багаторазових) введеннях
Матерiали реєстрацiйного досьє щодо дослiдження токсичностi при повторному (багаторазовому) введеннi мають бути спрямованим на виявлення будь-яких фiзiологiчних та/або паталогоанатомiчних змiн, що виникли внаслiдок багаторазового введення дiючої речовини або комбiнацiї дiючих речовин, та визначення того, як цi змiни залежать вiд дози.
У матерiалах реєстрацiйного досьє бажано наводити iнформацiю щодо двох дослiджень - короткострокового, тривалiстю 2 - 4 тижнi та довгострокового. Тривалiсть останнього залежить вiд тривалостi клiнiчного застосування лiкарського засобу. Його метою є експериментальне визначення i опис потенцiйних побiчних реакцiй, що мають бути врахованими при проведеннi клiнiчних випробувань;
в) генотоксичнiсть
У матерiалах реєстрацiйного досьє наводиться iнформацiя щодо мутагенного та кластогенного потенцiалу, метою якої є виявлення порушень, якi може спричинити дiюча речовина в генетичному матерiалi окремого органiзму або в клiтинах. Мутагеннi речовини є небезпечними для здоров'я людини, оскiльки дiя мутагену спричиняє мутацiї в статевих клiтинах i виникнення спадкових порушень, а також у соматичних клiтинах, що може призводити до розвитку злоякiсних новоутворень. Цi дослiдження є обов'язковими для всiх нових дiючих речовин;
г) канцерогеннiсть
У матерiалах реєстрацiйного досьє наводиться iнформацiя щодо дослiджень канцерогенного потенцiалу, якi зазвичай проводяться, якщо:
лiкарський засiб призначено для тривалого безперервного або перiодичного (з перервами) застосування протягом усього життя хворого;
при проведеннi токсикологiчних дослiджень токсичностi при повторному (багаторазовому) введеннi лiкарського засобу виявлено змiни, що викликають занепокоєння з приводу їхнього канцерогенного потенцiалу;
дiюча речовина належить до хiмiчного класу або є близькою за структурою до вiдомих канцерогенiв або коканцерогенiв.
Немає необхiдностi проводити такi дослiдження з безумовно генотоксичними сполуками, оскiльки вважається, що вони є трансвидовими канцерогенами i становлять небезпеку для людини;
ґ) репродуктивна токсичнiсть та токсичний вплив на розвиток потомства
У матерiалах реєстрацiйного досьє наводиться iнформацiя щодо дослiдження можливих порушень репродуктивної функцiї у чоловiкiв та жiнок, а також негативного впливу на потомство, якi здiйснюються за допомогою вiдповiдних випробувань. Вони включають дослiдження впливу на репродуктивну функцiю статевозрiлих самцiв i самиць, дослiдження токсичного та тератогенного впливу на потомство на всiх стадiях розвитку вiд зачаття до статевої зрiлостi, а також латентних ефектiв, коли дослiджуваний лiкарський засiб застосовувався для лiкування вагiтних самиць.
Вiдсутнiсть подiбних дослiджень у матерiалах реєстрацiйного досьє має бути вiдповiдним чином обґрунтовано.
У матерiалах реєстрацiйного досьє наводиться iнформацiя залежно вiд показань для застосування лiкарського засобу щодо проведення додаткових дослiджень (розвитку потомства) (за необхiдностi), наприклад, за наявностi обґрунтування введення лiкарського засобу статевонезрiлим тваринам.
Матерiали реєстрацiйного досьє щодо доклiнiчних дослiджень мають мiстити iнформацiю щодо дослiдження ембрiотоксичностi, якi проводяться, як правило, на двох видах ссавцiв, одним iз яких не є гризуни. Дослiдження пери- та постнатальної токсичностi має проводитися принаймнi на одному видi тварин. Якщо вiдомо, що метаболiзм лiкарського засобу для певного виду тварин є аналогiчним метаболiзму у людини, при проведеннi дослiджень доцiльно використовувати саме цей вид. Бажано також, щоб один з видiв тварин був тим самим, який використовувався при проведеннi дослiджень токсичностi при повторному (багаторазовому) введеннi;
д) мiсцева переносимiсть
Матерiали реєстрацiйного досьє щодо доклiнiчних дослiджень мають мiстити iнформацiю щодо мiсцевої переносимостi, метою якої є вивчення та визначення мiсцевої дiї лiкарського засобу (дiючих i допомiжних речовин) на тканини органiзму в тих дiлянках, що можуть контактувати з лiкарським засобом, внаслiдок його введення при клiнiчному застосуваннi.
Стратегiя дослiдження має бути спрямованою на те, щоб вiдрiзнити будь-який механiчний вплив введення або дiю, зумовлену фiзико-хiмiчними властивостями лiкарського засобу, вiд токсичного або фармакодинамiчного ефекту.
У матерiалах реєстрацiйного досьє має бути доведено, що дослiдження мiсцевої переносимостi здiйснювалися з використанням лiкарського засобу, розробленого для застосування людиною, пiд час якого тваринам контрольної групи вводиться наповнювач/розчинник для введення дослiджуваного лiкарського засобу та/або допомiжнi речовини. За необхiдностi слiд надати iнформацiю щодо додаткового включення групи позитивного контролю або речовини порiвняння.
Iнформацiя щодо дизайну дослiдження мiсцевої переносимостi (вибiр видiв тварин, тривалiсть, частота, спосiб введення, дози) повинна мiстити завдання дослiдження i рекомендованi умови клiнiчного застосування лiкарського засобу. За необхiдностi наводять iнформацiю щодо проведених дослiджень зворотностi мiсцевих ушкоджень.
Iнформацiю щодо дослiджень на тваринах можна замiнити iнформацiєю щодо випробувань з використанням валiдованих методiв in vitro, якщо результати дослiджень дозволяють визначити спiввiдношення користь/ризик.
Для хiмiчних речовин, що застосовуються мiсцево (наприклад, дермальнi, ректальнi, вагiнальнi), має наводитись оцiнка їх сенсибiлiзуючого потенцiалу з використанням щонайменше однiєї тест-системи (дослiдження на морських свинках або мiсцевих лiмфатичних вузлах).
5. Вимоги до матерiалiв реєстрацiйного досьє, наведених у Модулi 5 "Звiти про клiнiчнi випробування"
5.1. Модуль 5 має таку загальну схему:
змiст;
перелiк усiх клiнiчних випробувань у виглядi таблиць;
звiти про клiнiчнi випробування;
звiти про бiофармацевтичнi дослiдження;
звiти, що стосуються дослiдження фармакокiнетики при використаннi бiоматерiалiв людського походження;
звiти про фармакокiнетичнi дослiдження у людини;
звiти про фармакодинамiчнi дослiдження у людини;
звiти про дослiдження ефективностi та безпеки;
звiти про контрольованi клiнiчнi дослiдження щодо пiдтвердження заявлених показань для застосування;
звiти про неконтрольованi клiнiчнi дослiдження, звiти про аналiзи даних за кiлькома дослiдженнями i звiти про iншi клiнiчнi дослiдження;
звiти про пiсляреєстрацiйний досвiд застосування;
зразки iндивiдуальних реєстрацiйних форм та iндивiдуальнi списки пацiєнтiв;
посилання на джерела лiтератури.
5.2. Перелiк усiх клiнiчних випробувань у виглядi таблиць:
Особливу увагу необхiдно звернути на наявнiсть у матерiалах реєстрацiйного досьє iнформацiї щодо такого:
а) клiнiчна iнформацiя, яку необхiдно надати, повинна дозволити сформувати достатньо обґрунтованi й достовiрнi з наукової точки зору висновки щодо ефективностi та безпеки лiкарського засобу. Отже, важливою вимогою є те, що оприлюдненню пiдлягають результати всiх клiнiчних випробувань як сприятливi, так i несприятливi;
б) клiнiчним випробуванням завжди мають передувати вiдповiднi фармакологiчнi та токсикологiчнi дослiдження, проведенi на тваринах, iнформацiя щодо яких наведена у Модулi 4 матерiалiв реєстрацiйного досьє. Дослiдник повинен ознайомитися з висновками, зробленими в результатi фармакологiчних i токсикологiчних дослiджень, i тому заявник повинен надати йому принаймнi брошуру дослiдника, у яку повинна ввiйти вся вiдповiдна iнформацiя, вiдома на дату початку клiнiчних випробувань, включаючи хiмiчнi, фармакологiчнi та бiологiчнi данi, результати токсикологiчних, фармакокiнетичних i фармакодинамiчних дослiджень на тваринах, а також результати попереднiх клiнiчних випробувань, що надають адекватнi данi для обґрунтування характеру, масштабу та тривалостi планованого дослiдження; повнi звiти про фармакологiчнi та токсикологiчнi дослiдження повиннi надаватися на вимогу. Вiдносно матерiалiв людського або тваринного походження повиннi бути задiянi всi наявнi засоби щодо забезпечення безпеки у зв'язку з можливим поширенням збудникiв iнфекцiї до початку дослiдження;
в) власники реєстрацiйного посвiдчення повиннi забезпечити, щоб основна документацiя клiнiчного дослiдження (у тому числi iндивiдуальнi реєстрацiйнi форми) зберiгалася у власникiв отриманих результатiв, за винятком медичних карт стацiонарних/амбулаторних хворих (пацiєнтiв).
Медичнi карти стацiонарних/амбулаторних хворих (пацiєнтiв) повиннi зберiгатися у вiдповiдних умовах та протягом строку, передбаченого чинним законодавством.
Iнформацiя щодо будь-якої змiни права власностi стосовно наявних даних повинна бути вiдповiдно оформленою;
г) опис кожного клiнiчного дослiдження повинен мiстити достатню кiлькiсть iнформацiї, щоб скласти об'єктивний висновок про:
протокол, що мiстить обґрунтування, цiлi, статистичний метод i методологiю дослiдження з умовами його проведення та органiзацiї, i докладну iнформацiю про дослiджуваний лiкарський засiб;
сертифiкат про проходження аудиту (якщо такий проводився);
список дослiдникiв;
кожний дослiдник повинен повiдомити своє прiзвище, мiсце проживання, мiсце роботи, займану посаду, квалiфiкацiйнi данi та обов'язки при проведеннi клiнiчних дослiджень;
вказати, де проводилося дослiдження, надати iнформацiю щодо кожного окремого пацiєнта, включаючи iндивiдуальнi реєстрацiйнi форми;
заключний звiт, пiдписаний дослiдником, а при багатоцентровому дослiдженнi - всiма спiвдослiдниками або вiдповiдальним дослiдником;
ґ) повна документацiя щодо опису клiнiчного дослiдження, про яке йшлося вище, може надаватись на вимогу.
У матерiалах реєстрацiйного досьє має бути вiдображена думка дослiдника на основi експериментальних доказiв про безпеку лiкарського засобу за звичайних умов його застосування, його переносимiсть, ефективнiсть, про будь-яку корисну iнформацiю вiдносно показань для застосування та протипоказань, дозувань, тривалостi терапiї, а також щодо особливих запобiжних заходiв, яких необхiдно вжити пiд час лiкування та при появi клiнiчних симптомiв передозування. У звiтi про результати багатоцентрового дослiдження, який наводиться у матерiалах реєстрацiйного досьє, вiдповiдальний дослiдник у своїх висновках повинен висловити думку про безпеку та ефективнiсть дослiджуваного лiкарського засобу пiд час багатоцентрового дослiдження;
д) клiнiчнi спостереження щодо кожного дослiдження повиннi бути узагальненими iз зазначенням:
кiлькостi та статi пацiєнтiв, якi отримали лiкування;
вiдбору i розподiлу за вiком пацiєнтiв у дослiджуваних та контрольних групах;
кiлькостi пацiєнтiв, якi достроково вибули з дослiдження, i причин, з яких це вiдбулося;
якщо контрольованi дослiдження проводилися за зазначених вище умов, указати, що вiдбувалося з контрольною групою:
не одержувала лiкування,
одержувала плацебо,
одержувала iнший лiкарський засiб з вiдомою дiєю,
одержувала iнший вид лiкування без застосування лiкарських засобiв;
частоти побiчних реакцiй, що спостерiгалися;
подробиць щодо пацiєнтiв, що входять до груп пiдвищеного ризику, наприклад, люди лiтнього вiку, дiти, вагiтнi або жiнки репродуктивного вiку, або хворi, фiзiологiчний або патологiчний стан яких вимагає особливої уваги;
параметрiв або критерiїв оцiнки ефективностi та отриманих результатiв;
статистичної оцiнки результатiв, якщо цього вимагає дизайн дослiдження, i використаних змiнних факторiв.
У матерiалах реєстрацiйного досьє повинна вiдображатись iнформацiя щодо спостереження дослiдника про:
будь-якi ознаки звикання, залежностi або труднощiв у пацiєнтiв, що виникають при вiдмiнi лiкарського засобу;
будь-якi взаємодiї, що мали мiсце при одночасному введеннi з iншими лiкарськими засобами;
критерiї, що визначають необхiднiсть виключення деяких пацiєнтiв iз дослiдження;
фатальнi випадки пiд час дослiдження або в перiод подальшого спостереження.
Iнформацiя про нову комбiнацiю дiючих речовин повинна бути iдентичною даним про новий лiкарський засiб i до неї необхiдно включати обґрунтування безпеки та ефективностi комбiнацiї.
У разi повної або часткової вiдсутностi вищенаведених даних у матерiалах реєстрацiйного досьє необхiдно надати пояснення. За необхiдностi повинна надаватись iнформацiя про огляд та аналiз додаткових доклiнiчних токсикологiчних та фармакологiчних дослiджень у разi отримання при проведеннi клiнiчних дослiджень несподiваних результатiв.
Якщо лiкарський засiб призначено для тривалого застосування, у матерiалах реєстрацiйного досьє необхiдно надати опис будь-яких змiн фармакологiчної дiї в результатi багаторазового застосування лiкарського засобу, а також необхiдно обґрунтувати вибiр дозувань для тривалого застосування.
5.3. Звiти про клiнiчнi випробування:
5.3.1. Звiти про бiофармацевтичнi дослiдження:
До матерiалiв реєстрацiйного досьє необхiдно надати звiти про дослiдження бiодоступностi, порiвняльної бiодоступностi, бiоеквiвалентностi, кореляцiї in vitro - in vivo i опис бiоаналiтичних та аналiтичних методик.
Крiм того, за потреби демонстрацiї бiоеквiвалентностi лiкарських засобiв має бути наведена iнформацiя про проведену оцiнку їх бiодоступностi.
У разi застосування процедури бiовейвер до матерiалiв реєстрацiйного досьє необхiдно надати звiт про проведення дослiджень in vitro. Оцiнка та проведення дослiджень бiоеквiвалентностi або обґрунтування щодо його непроведення має бути надана вiдповiдно до вимог роздiлiв XIX та XX цього Порядку та керiвних настанов.
5.3.2. Звiти, що стосуються дослiдження фармакокiнетики при використаннi бiоматерiалiв людського походження:
До бiоматерiалiв людського походження вiдносять бiлки, клiтини, тканини й пов'язанi з ними матерiали, отриманi вiд людини, якi використовуються при проведеннi дослiджень in vitro або ex vivo з метою оцiнки фармакокiнетичних властивостей дiючих речовин.
У матерiалах реєстрацiйного досьє необхiдно надати звiти про дослiдження зв'язування з бiлками плазми, метаболiзму в печiнцi та взаємодiї дiючої речовини, а також дослiдження з використанням iнших бiоматерiалiв людського походження.
5.3.3. Звiти про фармакокiнетичнi дослiдження у людини:
а) у матерiалах реєстрацiйного досьє необхiдно описати такi фармакокiнетичнi характеристики:
абсорбцiя (швидкiсть i ступiнь);
розподiл;
метаболiзм;
виведення.
Необхiдно надати iнформацiю щодо опису клiнiчно важливих характеристик, включаючи значення кiнетичних даних при визначеннi схеми прийому лiкарського засобу для пацiєнтiв iз груп ризику, i вiдмiнностей мiж людиною та видами тварин, використаних при проведеннi доклiнiчних дослiджень.
У матерiалах реєстрацiйного досьє крiм iнформацiї щодо стандартних фармакокiнетичних дослiджень з використанням значної кiлькостi зразкiв при фармакокiнетичних аналiзах у популяцiї, що базуються на розрiдженому вiдборi проб пiд час клiнiчних дослiджень, також може наводитись iнформацiя щодо впливу внутрiшнiх i зовнiшнiх факторiв на варiабельнiсть взаємозв'язку мiж дозою i фармакокiнетичним вiдгуком. Необхiдно надати звiти про дослiдження фармакокiнетики та першi дослiдження переносимостi лiкарського засобу здоровими добровольцями та пацiєнтами, звiти про вивчення фармакокiнетики з метою оцiнки впливу внутрiшнiх i зовнiшнiх факторiв, а також звiти про фармакокiнетичнi дослiдження в популяцiї;
б) якщо лiкарський засiб зазвичай застосовується разом з iншими лiкарськими засобами, у матерiалах реєстрацiйного досьє має бути наданий опис дослiдження їх одночасного застосування, що проводилося для демонстрацiї можливої змiни фармакологiчної дiї.
Необхiдно надати iнформацiю щодо вивчення фармакокiнетичної взаємодiї дiючої речовини з iншими лiкарськими засобами або речовинами.
5.3.4. Звiти про фармакодинамiчнi дослiдження у людини:
а) у матерiалах реєстрацiйного досьє необхiдно пiдтвердити кореляцiю фармакодинамiчної дiї та ефективностi, включаючи:
взаємозв'язок доза - вiдгук та її розвиток у часi;
обґрунтування дозувань i способiв введення;
механiзм дiї, якщо можливо.
Необхiдно надати опис фармакодинамiчної дiї, не пов'язаний з ефективнiстю.
Iнформацiя щодо демонстрацiї фармакодинамiчної дiї в людини сама по собi є недостатньою для того, щоб зробити висновки стосовно будь-якої конкретної потенцiйної терапевтичної дiї;
б) якщо лiкарський засiб, зазвичай, застосовується разом з iншими лiкарськими продуктами, повинен бути наданий опис дослiдження їх одночасного застосування, що проводилося для демонстрацiї можливої змiни фармакологiчної дiї.
Необхiдно зазначити iнформацiю щодо вивчення фармакодинамiчної взаємодiї дiючої речовини з iншими лiкарськими засобами або речовинами.
5.3.5. Звiти про дослiдження ефективностi та безпеки:
5.3.5.1. Звiти про контрольованi клiнiчнi дослiдження щодо пiдтвердження заявлених показань для застосування:
Iнформацiя повинна бути надана щодо проведених клiнiчних дослiджень, якi за можливостi є рандомiзованими "контрольованими клiнiчними дослiдженнями", де дослiджуваний лiкарський засiб порiвнюється iз плацебо та вiдомим лiкарським засобом вiдомої терапевтичної ефективностi; використання будь-якого iншого дизайну дослiдження необхiдно обґрунтувати. Iнформацiя щодо лiкування контрольних груп буде рiзною у кожному конкретному випадку та буде залежати вiд етичних норм й галузi терапiї, тому в окремих випадках, можливо, буде наводитися iнформацiя щодо порiвняння ефективностi нового лiкарського засобу з ефективнiстю вiдомого лiкарського засобу вiдомої терапевтичної ефективностi, нiж з дiєю плацебо.
Пiд час надання оцiнки необхiдно застосовувати заходи, що дозволяють уникнути необ'єктивностi, включаючи методи рандомiзацiї та слiпого контролю.
Протокол дослiдження, що мiститься у матерiалах реєстрацiйного досьє, повинен включати докладний опис використаних статистичних методiв, число пацiєнтiв й причини для їхнього включення (включаючи розрахунки статистичної потужностi дослiджень), застосований рiвень значимостi та опис статистичної одиницi. Заходи, застосованi для уникнення необ'єктивної оцiнки, особливо методи рандомiзацiї, мають бути вiдповiдно обґрунтованi.
Iнформацiя щодо включення великої кiлькостi пацiєнтiв для участi в дослiдженнi не повинна вважатися рiвноцiнною замiною вiдповiдному контрольованому дослiдженню.
При наведеннi iнформацiї щодо аналiзу даних з безпеки необхiдно придiлити увагу обставинам, що призвели до змiни дози або необхiдностi супутнього застосування iншого лiкарського засобу, серйозних побiчних реакцiй, реакцiй, якi стали причиною виключення з участi в дослiдженнi та смертi. Необхiдно iдентифiкувати пацiєнтiв або групи пацiєнтiв дослiдження з пiдвищеним ступенем ризику й звернути особливу увагу на потенцiйно вразливi групи, кiлькiсть яких може бути невеликою, наприклад, дiти, вагiтнi, люди лiтнього вiку зi слабким здоров'ям, люди зi значними порушеннями обмiну речовин або екскрецiї та iн. Необхiдно придiлити увагу значенню оцiнки безпеки для можливих видiв застосування лiкарського засобу.
5.3.5.2. Звiти про неконтрольованi клiнiчнi дослiдження, звiти про аналiзи даних за кiлькома дослiдженнями i звiти про iншi клiнiчнi дослiдження:
Цi звiти необхiдно надати до матерiалiв реєстрацiйного досьє.
5.3.6. Звiти про пiсляреєстрацiйний досвiд застосування:
Якщо лiкарський засiб уже зареєстровано в iнших країнах, у матерiалах реєстрацiйного досьє необхiдно надати iнформацiю про побiчнi реакцiї на розглянутий лiкарський засiб та лiкарськi засоби з тiєю(ими) самою(ими) дiючою(ими) речовиною(ами), за можливостi, порiвняно з показниками їхнього застосування.
5.3.7. Зразки iндивiдуальних реєстрацiйних форм та iндивiдуальнi списки пацiєнтiв:
До матерiалiв реєстрацiйного досьє додаються зразки iндивiдуальних реєстрацiйних форм та списки пацiєнтiв зi збереженням конфiденцiйностi персональних даних пацiєнтiв даного дослiдження.
X. СПЕЦIАЛЬНI ВИМОГИ ДО МАТЕРIАЛIВ РЕЄСТРАЦIЙНОГО ДОСЬЄ (У ФОРМАТI ЗАГАЛЬНОГО ТЕХНIЧНОГО ДОКУМЕНТА) ЗА РIЗНИМИ ТИПАМИ ЗАЯВ
1. Спецiальнi вимоги до матерiалiв реєстрацiйного досьє для лiкарських засобiв з добре вивченим медичним застосуванням
Для лiкарських засобiв, АФI або дiючу(i) речовину(и) яких добре вивчено в медичному застосуваннi з пiдтвердженою ефективнiстю та прийнятним рiвнем безпеки, застосовуються такi спецiальнi вимоги до матерiалiв реєстрацiйного досьє.
Заявник повинен подавати Модулi 1, 2 i 3, як зазначено у роздiлi IX цього Порядку.
Для матерiалiв реєстрацiйного досьє Модулiв 4 i 5 у докладнiй науковiй бiблiографiї необхiдно вказувати доклiнiчнi та клiнiчнi характеристики лiкарського засобу.
Для пiдтвердження добре вивченого медичного застосування мають використовуватися спецiальнi вимоги:
а) фактори, якi необхiдно враховувати при визначеннi добре вивченого медичного застосування компонентiв лiкарських засобiв:
час, протягом якого використовується АФI або дiюча речовина,
кiлькiснi аспекти використання АФI або дiючої речовини,
ступiнь наукового iнтересу у використаннi АФI або дiючої речовини (з посиланням на опублiкованi науковi джерела),
послiдовнiсть наукових оцiнок.
Таким чином, для визначення добре вивченого застосування рiзних АФI або дiючих речовин може знадобитися оцiнка за рiзнi перiоди часу. Однак у будь-якому разi перiод часу, необхiдний для визначення добре вивченого медичного застосування АФI або дiючої речовини, має бути не меншим за 10 рокiв з часу першого систематичного та документованого використання цiєї АФI або дiючої речовини як лiкарського засобу;
б) матерiали реєстрацiйного досьє, наданi заявником, повиннi включати всi аспекти оцiнки безпеки та ефективностi, мiстити або давати посилання на огляд вiдповiдної лiтератури, з урахуванням перед- та пiсляреєстрацiйних дослiджень, та опублiкованої наукової лiтератури стосовно результатiв епiдемiологiчних дослiджень i, особливо, порiвняльних епiдемiологiчних дослiджень; всю документацiю, як позитивну, так i негативну. Щодо аспектiв про "добре вивчене медичне застосування" необхiдно уточнити, що "бiблiографiчне посилання" на iншi джерела доказiв (пiсляреєстрацiйнi дослiдження, епiдемiологiчнi дослiдження тощо), а не тiльки наявнi данi, що стосуються методiв контролю та випробувань, можуть бути вагомим доказом безпеки та ефективностi лiкарського засобу за умови, що в матерiалах реєстрацiйного досьє чiтко пояснено та обґрунтовано використання цих джерел iнформацiї;
в) особливу увагу необхiдно придiлити iнформацiї, якої не вистачає, та необхiдно обґрунтувати, чому може вважатися доведеним прийнятний рiвень безпеки та/або ефективностi, незважаючи на вiдсутнiсть деяких дослiджень;
г) у доклiнiчних та/або клiнiчних оглядах необхiдно пояснити значимiсть будь-яких поданих даних, що стосуються вже зареєстрованого лiкарського засобу, вiдмiнного вiд того, який пропонується до перереєстрацiї. Необхiдно надати обґрунтування з приводу того, чи можна заявлений лiкарський засiб уважати подiбним до вже зареєстрованого лiкарського засобу, незважаючи на iснуючi розбiжностi;
ґ) пiсляреєстрацiйний досвiд використання iнших лiкарських засобiв, що мiстять тi самi компоненти, є особливо важливим, i заявники пiд час надання матерiалiв реєстрацiйного досьє мають придiлити цьому питанню належну увагу.
2. Спецiальнi вимоги до матерiалiв реєстрацiйного досьє для генеричних лiкарських засобiв.
а) Матерiали реєстрацiйного досьє на генеричнi лiкарськi засоби повиннi мiстити данi, описанi в Модулях 1, 2 i 3 роздiлу IX цього Порядку за умови, що заявник має дозвiл власника реєстрацiйного посвiдчення оригiнального/референтного лiкарського засобу щодо посилання на Модулi 4 i 5 матерiалiв його реєстрацiйного досьє;
б) Матерiали реєстрацiйного досьє повиннi мiстити данi, описанi в Модулях 1, 2 i 3 роздiлу IX цього Порядку, а також данi, що доводять бiодоступнiсть i бiоеквiвалентнiсть з оригiнальним лiкарським засобом, за умови, що вiн не є бiологiчним лiкарським засобом.
Для цих лiкарських засобiв матерiали реєстрацiйного досьє щодо доклiнiчних/клiнiчних оглядiв/резюме (у разi їх проведення) мають включати такi елементи:
обґрунтування того, що лiкарський засiб є по сутi аналогiчним;
короткий опис домiшок, наявних у серiях АФI або дiючої(их) речовини(н), а також у готовому лiкарському засобi, який реєструється (у вiдповiдних випадках про продукти розпаду, що виникають при зберiганнi), разом з оцiнкою цих домiшок;
оцiнка дослiджень бiоеквiвалентностi або обґрунтування того, чому дослiдження не проводилися вiдповiдно до роздiлiв XIX та XX цього Порядку та керiвних настанов;
остання опублiкована лiтература, що є актуальною для АФI або дiючої речовини та цього реєстрацiйного досьє. Для цього можна використовувати анотацiї статей наукових журналiв;
кожне твердження в короткiй характеристицi лiкарського засобу, яке до цього не було вiдомо, або не витiкає, виходячи з властивостей лiкарського засобу та/або його терапевтичної групи, необхiдно розглянути в доклiнiчних/клiнiчних оглядах/резюме та пiдтвердити опублiкованою лiтературою та/або додатковими дослiдженнями. При цьому iнструкцiя для медичного застосування генеричного лiкарського засобу має мiстити iнформацiю, iдентичну тiй, яка наведена в iнструкцiї для медичного застосування референтного лiкарського засобу. Будь-якi вiдмiнностi iнструкцiї для медичного застосування генеричного лiкарського засобу мають обґрунтовуватись результатами, отриманими при багатоцентрових клiнiчних дослiдженнях;
якщо до складу генеричного лiкарського засобу входять солi, ефiри або похiднi зареєстрованої АФI або дiючої речовини, вiдмiннi вiд тих, що входять до складу референтного лiкарського засобу, заявник повинен надати додатковi данi з метою доведення еквiвалентностi характеристик безпеки та ефективностi;
якщо АФI або дiюча речовина генеричного лiкарського засобу має таку саму терапевтичну дiю, що й оригiнальний зареєстрований лiкарський засiб, але ця дiя пов'язана з iншою сiллю/ефiром/комплексом/похiдним, необхiдно пiдтвердити, що при цьому не змiнюється фармакокiнетика, фармакодинамiка та/або токсичнiсть тiєї частини речовини, яка є вiдповiдальною за цi властивостi, i може змiнити профiль безпеки/ефективностi. Якщо таких доказiв не iснує, то дану речовину можна вважати новою дiючою речовиною;
якщо лiкарський засiб призначено для iншого терапевтичного використання або представлено в iншiй лiкарськiй формi, або вiн має застосовуватися iншими шляхами введення, або в iнших дозах, або за iншої позологiї, необхiдно надати результати вiдповiдних токсикологiчних i фармакологiчних дослiджень та/або клiнiчних випробувань.
3. Спецiальнi вимоги до матерiалiв реєстрацiйного досьє для подiбних бiологiчних лiкарських засобiв (бiосимiлярiв)
У випадку реєстрацiї подiбних бiологiчних лiкарських засобiв, якi не можуть вважатись генеричними лiкарськими засобами через специфiчнiсть процесу виробництва, застосовуваної сировини, особливостей молекулярної будови та терапевтичної дiї, до матерiалiв реєстрацiйного досьє необхiдно надати додатковi данi, що пiдтверджують належний рiвень безпеки (токсикологiчнi чи iншi доклiнiчнi) та ефективностi (клiнiчнi).
При поданнi заяви на реєстрацiю подiбного бiологiчного лiкарського засобу пiсля закiнчення патентного захисту даних щодо зареєстрованого ранiше оригiнального бiологiчного лiкарського засобу необхiдно надавати такi матерiали:
iнформацiя, що подається, не повинна обмежуватися Модулями 1, 2 i 3 (фармацевтичнi, хiмiчнi та бiологiчнi данi), доповненими даними з бiоеквiвалентностi та бiодоступностi. Вид i кiлькiсть додаткових даних (тобто токсикологiчних, iнших доклiнiчних та вiдповiдних клiнiчних даних) мають визначатися у кожному окремому випадку згiдно з вiдповiдними вимогами та керiвництвами;
у зв'язку з рiзноманiтнiстю бiологiчних лiкарських засобiв може знадобитися надання iнформацiї щодо проведення iдентифiкувальних дослiджень, передбачених у Модулях 4 i 5 реєстрацiйного досьє, враховуючи спецiальну характеристику кожного конкретного лiкарського засобу.
У випадку, коли зареєстрований оригiнальний бiологiчний лiкарський засiб має бiльше одного показання до застосування, ефективнiсть i безпека подiбного бiологiчного лiкарського засобу повиннi пiдтверджуватися або за необхiдностi доводитися окремо для кожного iз заявлених показань.
4. Спецiальнi вимоги до матерiалiв реєстрацiйного досьє для лiкарських засобiв з фiксованою комбiнацiєю
Матерiали реєстрацiйного досьє повиннi стосуватися нових лiкарських засобiв, вироблених з використанням, принаймнi, двох вiдомих АФI або дiючих речовин, якi ранiше не застосовувалися у такiй комбiнацiї з терапевтичною метою.
Для таких лiкарських засобiв необхiдно надати матерiали повного реєстрацiйного досьє (Модулi 1 - 5).
За можливостi треба надати iнформацiю щодо дiльниць виробництва, допомiжних речовин та оцiнки безпеки.
5. Спецiальнi вимоги до матерiалiв реєстрацiйного досьє у виняткових випадках
Якщо заявник може довести, що для лiкарського засобу неможливо надати повнi данi про ефективнiсть i безпечнiсть за нормальних умов використання, оскiльки:
показання, для яких застосовується лiкарський засiб, зустрiчаються вкрай рiдко; у такий спосiб заявник не може зi зрозумiлих причин надати достатньо даних, або
iснуючi науковi данi не дають змоги надати повну iнформацiю, або
збiр такої iнформацiї буде суперечити загальноприйнятим принципам медичної етики,
то реєстрацiйне посвiдчення може видаватися з урахуванням певних зобов'язань.
Цi зобов'язання можуть включати таке:
заявник повинен здiйснити певну програму дослiджень у перiод часу, зазначений МОЗ, результати якої мають бути основою для повторної оцiнки спiввiдношення користь/ризик,
лiкарський засiб, що подається на реєстрацiю, має бути рецептурним та в деяких випадках застосовуватися тiльки пiд медичним наглядом, можливо в лiкарнi, а у випадку радiофармацевтичних препаратiв - квалiфiкованим спецiалiстом,
iнструкцiя для медичного застосування та будь-яка медична iнформацiя мають звертати увагу лiкаря на той факт, що наявний опис даного лiкарського засобу є певною мiрою неповним.
6. Спецiальнi вимоги до матерiалiв реєстрацiйного досьє за гiбридною заявою про реєстрацiю лiкарського засобу
У такому випадку заявник має подавати Модулi 1, 2 та 3 як зазначено в роздiлi IX цього Порядку.
Матерiали реєстрацiйного досьє для Модулiв 4 та/або 5 мають мiстити звiти про обмеженi доклiнiчнi дослiдження та/або клiнiчнi випробування, що проводилися заявником (залежно вiд певної ситуацiї щодо вiдмiнностей запропонованого лiкарського засобу та референтного препарату (пункт 6.5 роздiлу VI цього Порядку), та докладнi бiблiографiчнi данi щодо решти необхiдної iнформацiї для пiдтвердження безпеки та/або ефективностi запропонованого лiкарського засобу. Необхiдно надати обґрунтування з приводу того, чи можна запропонований лiкарський засiб уважати подiбним до вже зареєстрованого лiкарського засобу, незважаючи на iснуючi розбiжностi.
7. Спецiальнi вимоги до матерiалiв реєстрацiйного досьє для лiкарських засобiв з продукцiї in bulk
Для готових лiкарських засобiв, якi виготовленi за такими стадiями виробничого процесу як кiнцеве пакування та/або маркування, застосовуються особливi правила при поданнi матерiалiв реєстрацiйного досьє.
Заявник повинен подавати Модулi 1 та 2 матерiалiв реєстрацiйного досьє за вимогами, наведеними у роздiлi IX цього Порядку.
Для Модулiв 3, 4 i 5 фармацевтичнi, доклiнiчнi та клiнiчнi характеристики лiкарського засобу необхiдно вказувати у повному обсязi на пiдставi матерiалiв реєстрацiйного досьє виробника продукцiї in bulk. Вiдповiднi роздiли Модуля 3 також мають мiстити додатковi данi та документацiю заявника (власника реєстрацiйного посвiдчення на такий лiкарський засiб) щодо стадiй виробничого процесу, якi ним виконуються.
Крiм того, у вiдповiдних роздiлах матерiалiв реєстрацiйного досьє необхiдно чiтко вказувати найменування та мiсцезнаходження виробничих потужностей виробника продукцiї in bulk (який вiдповiдає за контроль якостi) та виробничих потужностей виробника, який вiдповiдає за випуск серiї. Iнформацiя щодо виробникiв має бути наведена у текстi маркування.
Необхiдно надати пiдтвердження реєстрацiї заявленого лiкарського засобу у споживчiй упаковцi в країнi виробника продукцiї in bulk.
8. Спецiальнi вимоги до матерiалiв реєстрацiйного досьє для бiологiчних лiкарських засобiв
8.1. Лiкарськi засоби, отриманi з кровi або плазми людини:
Положення Модуля 3 можуть частково не застосовуватися до лiкарських засобiв, отриманих з кровi або плазми людини, для яких матерiали реєстрацiйного досьє, оформленi згiдно з вимогами, викладеними в пунктах 3.2 та 3.3 глави 3 роздiлу IX цього Порядку, для вихiдних матерiалiв, отриманих з кровi або плазми людини, можуть бути замiненi мастер-файлом на плазму, оформленим вiдповiдно до цiєї частини.
Додатково до матерiалiв реєстрацiйного досьє може надаватись:
мастер-файл на плазму, що є окремим документом, який не входить до матерiалiв реєстрацiйного досьє, та мiстить всю вiдповiдну докладну iнформацiю про характеристики цiльної плазми людини, що використовується як вихiдний матерiал та/або сировина для виробництва субфракцiй/промiжних фракцiй, компонентiв допомiжних та дiючих речовин, якi є частиною лiкарського засобу;
iнформацiя щодо всiх закладiв охорони здоров'я/установ, якi займаються фракцiонуванням/обробкою плазми людини, має пiдготувати та поповнювати свiжими даними набiр вiдповiдної докладної iнформацiї, про яку йдеться у мастер-файлi на плазму;
якщо заявник не є власником мастер-файла на плазму, то власник повинен надати свiй мастер-файл заявнику для його подання. У будь-якому випадку заявник є вiдповiдальним за цей лiкарський засiб;
у будь-якому реєстрацiйному досьє на лiкарський засiб, що мiстить компоненти, отриманi з плазми людини, повинне бути посилання на мастер-файл на плазму, який вiдповiдає саме тiй плазмi, що використовувалася як вихiдний матерiал/сировина.
8.1.1. У мастер-файлi на плазму повинна мiститися iнформацiя про плазму, що використовувалася як вихiдний матерiал/сировина, а саме:
а) про походження плазми:
iнформацiя про заклади охорони здоров'я/установи, у яких проводиться вiдбiр кровi/плазми, уключаючи данi про проведенi iнспекцiї та отриману акредитацiю, а також епiдемiологiчнi данi про iнфекцiї, якi передаються через кров;
iнформацiя про заклади охорони здоров'я/установи, у яких проводиться дослiдження зразкiв донорської кровi i пулiв плазми, включаючи данi про проведенi iнспекцiї та отриману акредитацiю;
критерiї включення/виключення донорiв кровi/плазми;
прийнята система, що дозволяє вiдстежити шлях кожного донорського зразка вiд забору кровi/плазми до готового лiкарського засобу та навпаки;
б) про якiсть та безпеку плазми:
вiдповiднiсть монографiї Європейської фармакопеї або у разi її вiдсутностi iншiй визнанiй нацiональнiй фармакопеї;
аналiз зразкiв донорської кровi/плазми та пулiв на наявнiсть збудникiв iнфекцiй, уключаючи опис методiв аналiзу, та у випадку пулiв плазми данi з валiдацiї використаних методiв аналiзу;
технiчнi характеристики контейнерiв для вiдбору кровi та плазми, уключаючи iнформацiю про використанi розчини антикоагулянтiв;
умови зберiгання та транспортування плазми;
процедура з пiдтримки обладнання та/або перiод карантину;
характеристика пулу плазми.
8.1.2. У мастер-файлi на плазму повинна мiститися iнформацiя про прийняту систему взаємодiї мiж виробником одержаного з плазми лiкарського засобу, що здiйснює фракцiонування/обробку плазми, з одного боку, та/або закладами охорони здоров'я/установами, якi займаються вiдбором та аналiзом кровi/плазми, з iншого боку, яка визначає умови їхньої взаємодiї та погодження специфiкацiй.
8.1.3. Мастер-файл на плазму повинен також мiстити перелiк лiкарських засобiв, отриманих з даної плазми, незалежно вiд того, зареєстрованi цi лiкарськi засоби чи ще перебувають у процесi реєстрацiї, включаючи лiкарськi засоби, виготовленi для клiнiчних випробувань.
8.1.4. У мастер-файлi на плазму повинна мiститися iнформацiя про оцiнку та сертифiкацiю:
Для ще не зареєстрованих лiкарських засобiв заявник повинен надати матерiали повного реєстрацiйного досьє та окремо мастер-файл на плазму, якщо даний мастер-файл не проходив оцiнку ранiше.
Мастер-файл на плазму щорiчно поповнюється свiжими даними та повторно сертифiкується.
Мастер-файл на плазму мiстить iнформацiю щодо сертифiкацiї, повторної сертифiкацiї або змiни до мастер-файла на плазму, який вiдноситься до лiкарського засобу.
8.2. Вакцини:
Стосовно вакцин для людини, за умови застосування системи загального файла на вакцинний антиген (Vaccine Antigen Master File) (далi - ВАЗФ), положення Модуля 3 можуть частково не застосовуватись при оформленнi реєстрацiйного досьє згiдно з вимогами, викладеними у роздiлах 3.2 та 3.3 частини IX цього Порядку для вихiдних матерiалiв.
Матерiали реєстрацiйного досьє для вакцин, за винятком вакцин проти грипу людини, повиннi включати ВАЗФ на кожний антиген, який є активною речовиною цiєї вакцини.
8.2.1. ВАЗФ є частиною матерiалiв реєстрацiйного досьє, яка мiстить всю вiдповiдну детальну iнформацiю про бiологiчний, фармацевтичний та хiмiчний характер кожної з активних речовин, що входять до складу цього лiкарського засобу. Ця окрема частина може бути загальною для однiєї та бiльше моновалентних та/або комбiнованих вакцин, що представленi одним й тим самим заявником/власником реєстрацiйного посвiдчення.
Вакцина може мiстити один або декiлька рiзних антигенiв, кожен з яких є активною речовиною.
Комбiнована вакцина мiстить принаймнi два рiзних антигени, призначенi для профiлактики одного або декiлькох iнфекцiйних захворювань.
Моновалентна вакцина мiстить тiльки один антиген, призначений для профiлактики одного iнфекцiйного захворювання.
8.2.2. ВАЗФ повинен мiстити iнформацiю щодо якостi, про що йдеться у вiдповiдному роздiлi (дiюча речовина Модуля 3):
а) дiюча речовина:
загальна iнформацiя, включаючи вiдповiднiсть монографiям ДФУ, Європейської фармакопеї;
iнформацiя щодо виробництва дiючої речовини, включаючи iнформацiю про процес виробництва, вихiднi матерiали та сировину, особливi заходи з оцiнки ризику передачi збудникiв та додатковi заходи оцiнки безпеки споруд та обладнання;
характеристики дiючої речовини;
контроль якостi дiючої речовини;
стандартнi зразки речовин та матерiалiв;
система упаковка/укупорка для дiючої речовини;
стабiльнiсть дiючої речовини;
б) оцiнка та сертифiкацiя.
Для нових вакцин, якi мiстять новий вакцинний антиген, заявник повинен надати матерiали повного реєстрацiйного досьє, включаючи всi ВАЗФ, що належать до кожного окремого антигену, якi є частиною нової вакцини, якщо загальний файл на кожний окремий антиген не пройшов оцiнку ранiше.
Змiни до ВАЗФ на вже зареєстровану вакцину пiдлягають науковiй та технiчнiй оцiнцi. При позитивнiй оцiнцi на кожний файл видається сертифiкат вiдповiдностi, який супроводжується звiтом про проведену оцiнку.
Загальний файл вакцинного антигену має мiстити iнформацiю щодо сертифiкацiї, повторної сертифiкацiї або внесення змiн до загального файла на вакцинний антиген, який має вiдношення до лiкарського засобу.
9. Спецiальнi вимоги до матерiалiв реєстрацiйного досьє для радiофармацевтичних лiкарських засобiв та їх прекурсорiв
9.1. Радiофармацевтичнi лiкарськi засоби:
При реєстрацiї радiофармацевтичних лiкарських засобiв заяви потрiбно подавати разом з повними матерiалами реєстрацiйного досьє, якi повиннi мiстити такi специфiчнi особливостi:
9.1.1. У модулi 3:
а) у радiонуклiдному наборi, який пiсля постачання виробником має бути помiчений радiоактивною мiткою, дiючою речовиною вважається та частина лiкарської форми, яка зв'язується з радiонуклiдом або є носiєм радiонуклiда. Опис методу виробництва радiонуклiдних наборiв повинен включати подробицi виробництва набору та подробицi його рекомендованої кiнцевої обробки з метою створення радiофармацевтичного лiкарського засобу. Необхiднi специфiкацiї радiонуклiда повиннi (у вiдповiдних випадках) вiдповiдати загальнiй монографiї або спецiальним монографiям Європейської фармакопеї. Крiм того, має надаватись опис усiх сполук для мiчення радiоактивним iзотопом. Також необхiдно надати опис структури сполуки з радiоактивною мiткою.
Необхiдно надавати iнформацiю про радiонуклiди, якi задiянi в ядерних реакцiях.
Як первиннi, так i вториннi радiонуклiди вважаються активними речовинами генератора;
б) повиннi бути наданi докладнi данi про характер радiонуклiда, iдентичнiсть iзотопу, можливi домiшки, носiй, використання та специфiчну активнiсть;
в) до вихiдних матерiалiв належать матерiали, що є метою опромiнення;
г) необхiдно розглянути хiмiчну/радiофармацевтичну чистоту лiкарського засобу та її зв'язок з розподiлом в органiзмi людини;
ґ) необхiдно надати опис чистоти радiонуклiда, радiохiмiчної чистоти та специфiчної активностi;
д) для радiонуклiдних генераторiв необхiдно надати докладнi данi щодо випробувань первинних i вторинних радiонуклiдiв. Для генераторiв-елюатiв необхiдно надати методи випробування первинних радiонуклiдiв та iнших компонентiв генераторної системи;
е) вимога виражати вмiст дiючих речовин у перерахунку на масу активних компонентiв вiдноситься тiльки до радiонуклiдних наборiв. Для радiонуклiдiв радiоактивнiсть необхiдно виражати в бекерелях у конкретний день та за потреби у конкретний час з посиланням на часовий пояс. Має бути вказаний тип радiацiї;
є) для радiонуклiдних наборiв специфiкацiї готового лiкарського засобу повиннi включати випробування з визначення дiї лiкарських засобiв пiсля мiчення радiоактивним iзотопом. Мають бути включенi вiдповiднi засоби контролю радiохiмiчної та радiонуклiдної чистоти сполуки з радiоактивною мiткою. Потрiбно iдентифiкувати та проаналiзувати будь-який матерiал, необхiдний для мiчення радiоактивним iзотопом;
ж) має бути надана iнформацiя про стабiльнiсть радiонуклiдних генераторiв, радiонуклiдних наборiв i лiкарських засобiв з радiоактивною мiткою. Необхiдно документувати стабiльнiсть пiд час використання радiофармацевтичних лiкарських засобiв у флаконах, що мiстять декiлька доз.
9.1.2. У Модулi 4:
Матерiали реєстрацiйного досьє щодо оцiнки безпеки та ефективностi радiофармацевтичних лiкарських засобiв повиннi вiдповiдати вимогам, що висуваються до лiкарських засобiв та радiацiйної дозиметрiї. Необхiдно документувати рiвень впливу опромiнення на органи/тканини. Вiдповiдно до мiжнародної системи необхiдно зробити попереднiй розрахунок поглиненої дози випромiнювання при певному методi застосування.
9.1.3. У Модулi 5:
До матерiалiв реєстрацiйного досьє у вiдповiдних випадках необхiдно надати результати клiнiчних випробувань, в iнших - обґрунтувати показання до застосування за допомогою клiнiчних оглядiв.
9.2. Прекурсори радiонуклiдiв, що використовуються при виробництвi мiчених радiоактивними iзотопами лiкарських засобiв:
В особливих випадках, коли розглядається прекурсор радiонуклiда, який призначено тiльки для мiчення радiоактивним iзотопом, до матерiалiв реєстрацiйного досьє необхiдно надати iнформацiю, що стосується можливих наслiдкiв низької ефективностi мiчення або дисоцiацiї in vivo мiченого радiоактивним iзотопом кон'югата, тобто питань, пов'язаних з впливом вiльних радiонуклiдiв на хворого. Крiм того, має бути надана вiдповiдна iнформацiя, пов'язана з професiйною небезпекою, тобто впливом опромiнення на медичний персонал та довкiлля.
Зокрема, у вiдповiдних випадках повинна бути надана така iнформацiя:
9.2.1. У Модулi 3:
Як визначено у пiдпунктi 9.1.1 пункту 9.1 цього роздiлу, у вiдповiдних випадках положення Модуля 3 належать до реєстрацiї прекурсорiв радiонуклiдiв.
9.2.2. У Модулi 4:
До матерiалiв реєстрацiйного досьє мають бути наданi результати дослiджень щодо токсичностi одноразової та багаторазової дози, проведених вiдповiдно до принципiв належної лабораторної практики, якщо iнше обґрунтування вiдсутнє.
У цьому випадку надання iнформацiї про дослiдження мутагенностi радiонуклiда не вважається достатнiми.
Повинна бути надана iнформацiя про хiмiчну токсичнiсть та розподiл вiдповiдного "холодного" нуклiда.
9.2.3. У Модулi 5:
До матерiалiв реєстрацiйного досьє необхiдно надати iнформацiю, що демонструє клiнiчне застосування прекурсору радiонуклiда, приєднаного до вiдповiдних молекул, оскiльки у випадку, коли розглядається прекурсор радiонуклiда, призначений тiльки для мiчення радiоактивним iзотопом дослiджуваного лiкарського засобу, клiнiчнi данi, отриманi в дослiдженнях самого прекурсору, не мають значення.
10. Спецiальнi вимоги до матерiалiв реєстрацiйного досьє для гомеопатичних лiкарських засобiв
Ця глава стосується особливих вимог до оформлення Модулiв 3 i 4 матерiалiв реєстрацiйного досьє на гомеопатичнi лiкарськi засоби, якi визначенi в абзацах сiмнадцятому та вiсiмнадцятому роздiлу II цього Порядку.
10.1. У Модулi 3:
Спецiальнi вимоги до Модуля 3 матерiалiв реєстрацiйного досьє для гомеопатичних лiкарських засобiв:
а) термiнологiя
Латинська назва гомеопатичної сировини, описаної у матерiалах реєстрацiйного досьє, повинна вiдповiдати латинськiй назвi, зазначенiй в Європейськiй фармакопеї, або в разi вiдсутностi - Нiмецькiй гомеопатичнiй фармакопеї (GHP), Гомеопатичнiй фармакопеї США (HPUS), Британськiй гомеопатичнiй фармакопеї (BHP), Гомеопатичнiй фармакопеї Швабе). У вiдповiдних випадках необхiдно надати традицiйно використовувану назву;
б) контроль вихiдних матерiалiв
Опис та данi щодо вихiдних матерiалiв, тобто щодо всiх використаних матерiалiв, уключаючи сировину та промiжнi матерiали, до кiнцевого розведення у готовому лiкарському засобi повиннi бути пiдкрiпленi додатковими даними про гомеопатичну сировину.
Загальнi вимоги до якостi стосуються всiх вихiдних матерiалiв i сировини, а також промiжних етапiв виробничого процесу до кiнцевого розведення у готовому лiкарському засобi. Якщо можна, потрiбно провести дослiдження у разi присутностi токсичних компонентiв, а також тодi, коли якiсть неможливо контролювати при кiнцевому розведеннi через високий ступiнь розведення. Необхiдно надати повний опис кожної стадiї виробничого процесу, починаючи вiд вихiдних матерiалiв до кiнцевого розведення у готовому лiкарському засобi.
Якщо застосовуються декiлька розведень, цi стадiї розведення необхiдно проводити вiдповiдно до методiв виробництва гомеопатичних засобiв, якi викладено у вiдповiднiй монографiї Європейської фармакопеї, або, за її вiдсутностi, в iншiй фармакопеї;
в) методи контролю готового лiкарського засобу
До готових гомеопатичних лiкарських засобiв застосовуються загальнi вимоги до якостi. Будь-якi виключення вимагають ретельного обґрунтування заявником.
Необхiдно iдентифiкувати та проаналiзувати всi компоненти, якi мають потенцiйну токсичнiсть. Якщо неможливо iдентифiкувати та проаналiзувати всi компоненти, якi можуть виявитися токсичними, наприклад через їх високий ступiнь розведення в готовому лiкарському засобi, потрiбно обґрунтувати якiсть лiкарського засобу за допомогою повної валiдацiї виробничого процесу та процесу розведення;
г) випробування на стабiльнiсть
Необхiдно пiдтвердити стабiльнiсть готового лiкарського засобу. Данi щодо стабiльностi гомеопатичного лiкарського засобу, як правило, дiйснi як для розчинiв, так i для порошкiв, з яких був вироблений даний лiкарський засiб. Якщо iдентифiкацiя або аналiз АФI або дiючої речовини неможливi через високий ступiнь розведення, можуть розглядатися данi щодо стабiльностi лiкарського засобу.
10.2. У Модулi 4:
Спецiальнi вимоги до Модуля 4 матерiалiв реєстрацiйного досьє для гомеопатичних лiкарських засобiв:
Необхiдно обґрунтувати вiдсутнiсть будь-якої iнформацiї, наприклад, має надаватися обґрунтування, яким чином може доводитися прийнятний рiвень безпеки, незважаючи на те, що вiдсутнi деякi дослiдження.
11. Спецiальнi вимоги до матерiалiв реєстрацiйного досьє для рослинних лiкарських засобiв
До заяви на реєстрацiю/перереєстрацiю рослинних лiкарських засобiв додаються матерiали повного реєстрацiйного досьє, до якого повиннi бути включенi такi специфiчнi данi.
Спецiальнi вимоги до Модуля 3 матерiалiв реєстрацiйного досьє для рослинних лiкарських засобiв:
У Модулi 3:
Положення Модуля 3, включаючи вiдповiднiсть монографiї Європейської фармакопеї, стосуються реєстрацiї/перереєстрацiї готових рослинних лiкарських засобiв. При цьому береться до уваги рiвень наукових знань на момент подання заяви.
Стосовно рослинних лiкарських засобiв розглядаються такi специфiчнi аспекти:
а) рослиннi субстанцiї та рослиннi препарати
Щодо номенклатури рослинної субстанцiї необхiдно вказати ботанiчнi характеристики рослини (рiд, вид, рiзновид та автора), хемотип (у вiдповiдних випадках) частин рослини, iншi назви (синонiми, зазначенi в iнших фармакопеях) та лабораторний код.
Щодо номенклатури рослинного препарату необхiдно вказати ботанiчнi характеристики рослини (рiд, вид, рiзновид та автора), хемотип (у вiдповiдних випадках) частин рослини, визначення препарату, спiввiдношення рослинної субстанцiї до рослинного препарату, розчинник(и), iншi назви (синонiми, зазначенi в iнших фармакопеях) та лабораторний код.
При пiдготовцi документiв щодо структури рослинної субстанцiї та рослинного препарату (у вiдповiдних випадках) необхiдно надати iнформацiю про фiзичну форму, опис компонентiв з вiдомою терапевтичною дiєю або маркерiв (молекулярну формулу, вiдносну молекулярну масу, структурну формулу, уключаючи вiдносну та абсолютну стереохiмiю), а також iнформацiю про iншi компоненти.
При пiдготовцi документiв щодо виробництва рослинної субстанцiї у вiдповiдних випадках необхiдно надати iнформацiю про кожного постачальника сировини, включаючи пiдрядникiв, а також iнформацiю про кожну передбачену виробничу дiльницю або устаткування, задiяне при виробництвi/заготовцi та контролi рослинної субстанцiї.
При пiдготовцi документiв щодо виробництва рослинного препарату у вiдповiдних випадках необхiдно вказати iнформацiю про кожного постачальника сировини, уключаючи пiдрядникiв, а також iнформацiю про кожну передбачену виробничу дiльницю або устаткування, задiяне при виробництвi та контролi рослинного засобу.
Щодо опису виробничого процесу рослинної субстанцiї необхiдно надати iнформацiю, у якiй буде вiдповiдним чином описано виробництво субстанцiї та заготiвлю рослинної сировини, уключаючи географiчне мiсце вирощування та культивування лiкарської рослини, умови її збору, висушування та зберiгання.
Щодо опису виробничого процесу та процедури контролю рослинного препарату необхiдно надати iнформацiю, у якiй буде вiдповiдним чином описане виробництво засобу, включаючи опис обробки сировини, розчинникiв i реагентiв, стадiї очищення та стандартизацiї.
Щодо розробки виробничого процесу необхiдно надати (у вiдповiдних випадках) короткий опис розробки рослинної субстанцiї та рослинного препарату з урахуванням передбачуваного шляху застосування та показань. У вiдповiдних випадках має надаватися обговорення результатiв порiвняння фiтохiмiчної будови рослинної субстанцiї та рослинного препарату, що використовувались на пiдтримку бiблiографiчних даних, та рослинної субстанцiї i рослинного препарату, що є дiючою речовиною готового рослинного лiкарського засобу, на який подано заяву про державну перереєстрацiю.
При описi структури та iнших характеристик рослинної субстанцiї за необхiдностi має надаватися iнформацiя про ботанiчнi, макро- та мiкроскопiчнi, фiтохiмiчнi характеристики i бiологiчну активнiсть.
При описi структури та iнших характеристик рослинного препарату у разi необхiдностi має надаватися iнформацiя про фiтохiмiчнi i фiзико-хiмiчнi характеристики та бiологiчну активнiсть.
За необхiдностi мають надаватися специфiкацiї для рослинної субстанцiї та рослинного препарату.
У вiдповiдних випадках потрiбно надати опис аналiтичних методiв, що використовувалися при випробуваннi рослинної субстанцiї та рослинного препарату.
При описi валiдацiї аналiтичних методик у вiдповiдних випадках потрiбно надати iнформацiю про валiдацiю методик, включаючи експериментальнi данi для методiв аналiзу, якi використовуються при випробуваннях рослинної субстанцiї та рослинного препарату.
При описi аналiзу серiй у вiдповiдних випадках потрiбно надати опис та результати аналiзу серiй рослинних субстанцiй та рослинних препаратiв у такому виглядi, як монографiї фармакопей.
У вiдповiдних випадках потрiбно надати обґрунтування специфiкацiй рослинних субстанцiй i рослинних препаратiв.
У вiдповiдних випадках потрiбно надати iнформацiю про стандартнi зразки або стандартнi препарати, що використовуються при випробуваннях рослинної субстанцiї та рослинного препарату;
б) готовi рослиннi лiкарськi засоби
Короткий опис розробки складу лiкарського засобу повинен уключати iнформацiю про розробку готового рослинного лiкарського засобу з увагою на передбачуваний спосiб застосування та показання. У вiдповiдних випадках потрiбно надати вiдомостi про результати порiвняння фiтохiмiчної будови лiкарського засобу, який використовується на пiдтримку бiблiографiчних даних, та лiкарського засобу, на який подано заяву.
12. Спецiальнi вимоги до матерiалiв реєстрацiйного досьє для лiкарських засобiв прогресивної терапiї (високотехнологiчнi препарати)
12.1. При поданнi заяви на реєстрацiю лiкарських засобiв прогресивної терапiї (високотехнологiчнi препарати) необхiдно дотримуватися вимог до матерiалiв реєстрацiйного досьє (Модулi 1 - 5), як зазначено у роздiлi IX цього Порядку.
Для бiологiчних лiкарських засобiв необхiдно застосовувати спецiальнi вимоги до матерiалiв реєстрацiйного досьє для Модулiв 3, 4 та 5, як зазначено у главi 8 цього роздiлу. Додатково спецiальнi вимоги до матерiалiв реєстрацiйного досьє на лiкарськi засоби прогресивної терапiї (високотехнологiчнi препарати) описанi у пунктах 12.2, 12.3 та 12.4 цiєї глави цього роздiлу. За необхiдностi, враховуючи особливостi лiкарських засобiв прогресивної терапiї (високотехнологiчнi препарати), до матерiалiв реєстрацiйного досьє надається додаткова iнформацiя.
Через специфiчну природу лiкарських засобiв прогресивної терапiї (високотехнологiчних препаратiв) можуть бути наданi матерiали, що складенi вiдповiдно до наукових керiвництв з ефективностi, безпеки та якостi високотехнологiчних препаратiв на основi оцiнки ризику для визначення ступеня якостi, доклiнiчнi та клiнiчнi данi, якi будуть включенi до матерiалiв реєстрацiйного досьє.
Матерiали реєстрацiйного досьє повиннi включати аналiз ризикiв, що може охоплювати всю розробку. Iнформацiя про фактори ризику, якi можуть розглядатися, повинна вiдображати: походження клiтин (аутологiчнi, галогеннi, ксеногеннi), здатнiсть до розростання та/або видозмiнювання та iнiцiювання iмунної вiдповiдi; манiпуляцiї на клiтинному рiвнi; комбiнацiя клiтин з бiоактивними молекулами або структурними матерiалами; природу лiкарського засобу генної терапiї; ступiнь здатностi до реплiкацiї вiрусiв або мiкроорганiзмiв, що використовуються in vivo; рiвень iнтеграцiї нуклеїнових кислот або генiв у геном; тривала функцiональнiсть; ризик онкогенностi та спосiб введення або застосування.
Матерiали реєстрацiйного досьє щодо аналiзу ризикiв можуть мiстити iнформацiю про вiдповiднi доклiнiчнi та клiнiчнi данi або досвiд застосування iнших високотехнологiчних препаратiв.
Будь-яке вiдхилення вiд вимог цього пункту має бути науково обґрунтовано у Модулi 2. Матерiали реєстрацiйного досьє Модуля 2 також можуть включати iнформацiю щодо аналiзу ризикiв, що описаний вище, у разi застосування. У такому випадку має бути оцiнено використовувану методику, природу певних ризикiв та результати пiдходу на пiдставi оцiнки ризику для програми з розробки та оцiнки, а також має бути наданий опис будь-яких вiдхилень вiд вимог цього пункту внаслiдок аналiзу ризикiв.
12.2. Спецiальнi вимоги до Модуля 3:
12.2.1. Спецiальнi вимоги для усiх лiкарських засобiв прогресивної терапiї (високотехнологiчнi препарати):
У матерiалах реєстрацiйного досьє необхiдно надати опис системи простежуваностi, яку заявник буде встановлювати та пiдтримувати для гарантiї того, що окремий продукт та його вихiднi матерiали i сировина, включаючи усi речовини, що вступають у контакт з клiтинами i тканинами, що можуть мiститися у препаратi, можуть бути простеженi за допомогою встановлення походження, виробництва, упаковки, зберiгання, транспортування та доставки у заклад охорони здоров'я/установу.
Система простежуваностi щодо людських клiтин та тканин, а також клiтин кровi має вiдповiдати вимогам, встановленим у вiдповiдних керiвництвах.
12.2.2. Спецiальнi вимоги для лiкарських засобiв генної терапiї:
Лiкарський засiб генної терапiї - бiологiчний лiкарський засiб, якому властивi такi характеристики:
дiюча речовина такого лiкарського засобу мiстить або складається з рекомбiнантної нуклеїнової кислоти, що використовується або вводиться людинi з метою впорядкування, вiдновлення, замiни, додавання або вилучення генетичної послiдовностi;
його терапевтична, профiлактична або дiагностична дiя пов'язана безпосередньо з послiдовнiстю рекомбiнантної нуклеїнової кислоти, яку вiн мiстить, або з продуктом генетичної експресiї цiєї послiдовностi.
До лiкарських засобiв генної терапiї не належать вакцини для профiлактики iнфекцiйних захворювань.
Лiкарськi засоби генної терапiї включають:
чисту нуклеїнову кислоту;
складну нуклеїнову кислоту або невiруснi вектори;
вiруснi вектори;
генетично модифiкованi клiтини.
У матерiалах реєстрацiйного досьє для лiкарських засобiв генної терапiї повинно бути вiдображено iнформацiю про три основнi елементи виробничого процесу:
вихiднi матерiали - матерiали, з яких виробляється дiюча речовина, такi як, наприклад, ген, що становить iнтерес, плазмiди експресiї, банки клiтин та запаси вiрусiв або невiрусний вектор;
АФI або дiюча речовина - рекомбiнантний вектор, вiрус, чистi або складнi плазмiди, клiтини, що продукують вiрус, генетично модифiкованi in vitro клiтини;
готовий лiкарський засiб - дiюча речовина в її кiнцевiй безпосереднiй упаковцi для призначеного медичного застосування. Залежно вiд типу лiкарського засобу прогресивної терапiї (високотехнологiчного препарату), способу та умов його застосування може бути потрiбна обробка клiтин пацiєнта ex vivo.
До матерiалiв реєстрацiйного досьє у Модулi 3 необхiдно надати:
а) iнформацiю про вiдповiднi характеристики лiкарського засобу генної терапiї, включаючи його експресiю в популяцiї цiльових клiтин, а також iнформацiю щодо джерела, структури, характеристик та визначення справжностi кодуючої послiдовностi гена, включаючи її цiлiснiсть i стабiльнiсть. Крiм терапевтичного гена необхiдно надати iнформацiю про повну послiдовнiсть iнших генiв, регуляторнi елементи та скелет вектора;
б) iнформацiю щодо характеристик вектора, який використовувався для переносу та доставки гена. Вона повинна включати фiзико-хiмiчнi та/або бiологiчно-iмунологiчнi характеристики.
Для лiкарських засобiв, в яких використано мiкроорганiзми (наприклад бактерiї або вiруси) для переносу гена (бiологiчний перенос гена), необхiдно надати iнформацiю про патогеннiсть вихiдного штаму та про його тропнiсть до певних тканин i видiв клiтин, а також про залежнiсть взаємодiї вiд клiтинного циклу.
Для лiкарських засобiв, в яких використано небiологiчнi засоби переносу гена, необхiдно надати iнформацiю про фiзико-хiмiчнi властивостi окремих компонентiв та всiєї комбiнацiї;
в) опис принципiв створення або пiдтримки банку клiтин та їх характеристик, якi належать до лiкарських засобiв, що переносять ген;
г) iнформацiю про джерело клiтин, що мiстять рекомбiнантний вектор.
Якщо джерелом клiтин є людина, то необхiдно надати iнформацiю про її вiк, стать, результати мiкробiологiчного та вiрусного дослiджень, критерiї виключення та країну проживання.
Якщо джерелом клiтин є тварина, то необхiдно надавати докладну iнформацiю щодо:
джерела тварин;
утримання тварин та догляду за ними;
трансгенних тварин (методи створення, характеристика трансгенних клiтин, характер введеного гена);
заходiв щодо запобiгання та контролю iнфекцiй у тваринних донорiв;
тестування на наявнiсть збудникiв iнфекцiй;
примiщень;
контролю вихiдних матерiалiв i сировини.
Необхiдно надати опис методу збору клiтин, включаючи мiсце, тип тканини, робочий процес, транспортування, зберiгання та системи вiдстеження, а також процедури контролю пiд час збору клiтин;
ґ) оцiнку вiрусної безпеки, а також системи вiдстеження лiкарських засобiв вiд донора до готового лiкарського засобу, що є важливою частиною матерiалiв реєстрацiйного досьє, що надається. Наприклад, має бути виключена присутнiсть вiрусу, що здатний до реплiкацiї, у джерелах нереплiкацiйних компетентних вiрусних векторiв.
12.2.3. Спецiальнi вимоги до лiкарських засобiв соматоклiтинної терапiї:
Лiкарський засiб соматоклiтинної терапiї - бiологiчний лiкарський засiб, якому властивi такi характеристики:
мiстить чи складається з клiтин або тканин, бiологiчнi характеристики, фiзiологiчнi функцiї або структурнi властивостi яких є важливими для клiнiчного застосування, були змiненi у результатi значних манiпуляцiй або з клiтин чи тканин, якi не призначенi для використання з тiєю самою метою у донора або реципiєнта;
має властивостi для, або використовується у, або вводиться людинi з метою лiкування, профiлактики або дiагностики захворювання шляхом фармакологiчної, iмунологiчної або метаболiчної дiї його клiтин або тканин.
Лiкарськi засоби соматоклiтинної терапiї включають:
клiтини, якi за допомогою манiпуляцiй змiнили якiснi та кiлькiснi параметри своїх iмунологiчних, метаболiчних або iнших функцiональних властивостей;
клiтини сортованi, вiдiбранi, з якими проведено вiдповiднi манiпуляцiї та якi надалi будуть задiянi у виробничому процесi при отриманнi готового лiкарського засобу;
манiпульованi клiтини та поєднанi з неклiтинними компонентами (наприклад, бiологiчними або неактивними матрицями або медичними засобами), якi чинять основну призначену дiю в готовому лiкарському засобi;
похiднi аутологiчних клiтин, експресованих in vitro за специфiчних культуральних умов;
генетично модифiкованi клiтини або клiтини, з якими проводилися iншi манiпуляцiї, щоб експресувати ранiше неекспресованi гомологiчнi або негомологiчнi функцiональнi властивостi.
Весь виробничий процес, вiд збору клiтин у пацiєнта (аутологiчна ситуацiя) до введення йому лiкарського засобу, вважається одним втручанням.
У матерiалах реєстрацiйного досьє для лiкарських засобiв соматоклiтинної терапiї повинно бути вiдображено iнформацiю про три елементи виробничого процесу:
вихiднi матерiали - матерiали, з яких виробляється дiюча речовина, тобто органи, тканини, рiдини органiзму або клiтини;
АФI або дiюча речовина - манiпульованi клiтини, клiтиннi лiзати, пролiферувальнi клiтини та клiтини, що використовуються у сполученнi з неактивними матрицями та медичними засобами;
готовий лiкарський засiб - дiюча речовина, пiдготовлена у її кiнцевiй безпосереднiй упаковцi для призначеного медичного застосування.
У матерiалах реєстрацiйного досьє у Модулi 3 має бути представлена:
а) загальна iнформацiя про дiючу речовину
Дiючi речовини лiкарських засобiв клiтинної терапiї складаються з клiтин, якi внаслiдок обробки in vitro проявляють профiлактичнi, дiагностичнi або терапевтичнi властивостi, вiдмiннi вiд їх первинних фiзiологiчних i бiологiчних властивостей.
Необхiдно надати опис типу клiтин та використаної культури. Має бути надана документацiя на тканини, органи або бiологiчнi рiдини, з яких отримано клiтини, а також аутологiчний, алогенний або ксеногенний характер донорських зразкiв та їх географiчне походження. Необхiдно надати детальний опис заготiвлi клiтин, зразкiв та умов їх зберiгання до подальшої обробки. Для алогенних клiтин особливу увагу необхiдно придiлити найпершiй стадiї процесу - вiдбору донорiв. Має бути надана iнформацiя про тип проведених манiпуляцiй i фiзiологiчну функцiю клiтин, якi використовуються як дiюча речовина;
б) iнформацiя щодо вихiдних матерiалiв для дiючої речовини.
12.2.3.1. Соматичнi клiтини людини:
Лiкарськi засоби соматоклiтинної терапiї з використанням клiтин людини виробляються з певної кiлькостi (пулу) життєздатних клiтин, якi отримують у результатi виробничого процесу, що починається на рiвнi органiв або тканин, отриманих вiд людини, або на рiвнi чiтко визначеної системи банку клiтин, де клiтинний пул належить до безперервних клiтинних лiнiй. У рамках цього роздiлу дiючою речовиною вважається посiяний пул клiтин людини, а готовим лiкарським засобом вважається пул клiтин людини, виготовлених для призначеного медичного застосування. У матерiалах реєстрацiйного досьє необхiдно надати повну iнформацiю про вихiднi матерiали та кожну стадiю виробничого процесу, включаючи питання вiрусної безпеки.
У матерiалах реєстрацiйного досьє необхiдно надати iнформацiю про такi характеристики донора, як вiк, стать, мiкробiологiчний статус, критерiї виключення та країну проживання; опис вiдбору зразкiв, включаючи мiсце, тип, робочий процес створення пулу, перевезення; зберiгання та монiторинг; методи контролю вiдбору зразкiв.
Окремi вимоги, викладенi для соматичних клiтин людини, стосуються iнформацiї щодо пiдготовки та контролю якостi систем створення банку клiтин. Цi особливостi належать до алогенних i ксеногенних клiтин.
У матерiалах реєстрацiйного досьє необхiдно надати iнформацiю про використання будь-якої сировини (наприклад, цитокинiв, факторiв росту або культурального середовища) або можливих допомiжних речовин i медичних засобiв (наприклад, засоби для сортування клiтин, бiосумiснi полiмери, матрицi, волокна, гранули) з точки зору їх бiосумiсностi, функцiональностi, а також ризику передачi збудникiв iнфекцiї.
12.2.3.2. Твариннi соматичнi клiтини (ксеногеннi):
У матерiалах реєстрацiйного досьє необхiдно надавати докладну iнформацiю щодо таких питань:
джерела тварин;
тримання та догляд за тваринами;
генетично модифiкованi тварини (методи створення, характеристики трансгенних клiтин, характер введеного або вилученого нокаутного гена);
заходи щодо запобiгання та контролю iнфекцiй у тваринних донорiв;
тестування на наявнiсть збудникiв iнфекцiї, включаючи мiкроорганiзми, що передаються вертикально (також ендогеннi ретровiруси);
устаткування;
системи створення банку клiтин;
контроль вихiдних матерiалiв i сировини.
У матерiалах реєстрацiйного досьє у Модулi 3 має бути представлена:
а) iнформацiя про процес виробництва дiючої речовини i готового лiкарського засобу - необхiдно надати iнформацiю про рiзнi стадiї виробничого процесу такi, як, наприклад, вiддiлення органа/тканини, вибiр популяцiї клiтин, що становить iнтерес, культивування клiтин in vitro, трансформацiя клiтин за допомогою фiзико-хiмiчних агентiв або шляхом переносу гена;
б) характеристика дiючої речовини - необхiдно надати всю iнформацiю, яка стосується цього пункту, про характеристики популяцiї клiтин, що становлять iнтерес, включаючи її iдентичнiсть (оригiнальнi види, бендiнгова цитогенетика (banding cytogenetics), морфологiчний аналiз), чистоту (побiчнi мiкробнi агенти i клiтиннi забруднювачi), силу дiї (певна бiологiчна активнiсть) та придатнiсть (аналiз карiотипу та онкогенностi) для призначеного медичного застосування;
в) фармацевтична розробка готового лiкарського засобу - крiм iнформацiї про специфiчний спосiб введення (внутрiшньовенна iнфузiя, локальна iн'єкцiя, хiрургiчна трансплантацiя) має бути також надана iнформацiя про можливе використання допомiжних медичних засобiв (бiосумiсних полiмерiв, матриць, волокон, гранул) з точки зору їх бiосумiсностi та зносостiйкостi;
г) система простежування - необхiдно надати докладну схему процесу, який забезпечує систему простежування препаратiв вiд донора до готового лiкарського засобу.
12.2.4. Спецiальнi вимоги до лiкарських засобiв прогресивної терапiї (високотехнологiчнi препарати), що мiстять пристрої для введення:
У матерiалах реєстрацiйного досьє необхiдно надати опис фiзичних характеристик та дiї продукту, а також опис методiв проектування продукту.
Необхiдно описати взаємодiю та сполучуванiсть генiв, клiтин та/або тканин i структурних компонентiв пристрою.
Пристрiй для введення або активний медичний вирiб, що iмплантується, можуть бути невiд'ємною частиною дiючої речовини. Якщо пристрiй для введення або активний медичний вирiб, що iмплантується, комбiнується з клiтинами пiд час виробництва, застосування або введення готових продуктiв, вони повиннi розглядатися як невiд'ємна частина готового лiкарського засобу.
У матерiалах реєстрацiйного досьє необхiдно надати iнформацiю, що стосується пристрою для введення або активного медичного виробу, що iмплантується (що є невiд'ємною частиною дiючої речовини або готового лiкарського засобу), та є необхiдною для оцiнки комбiнованого високотехнологiчного препарату. Така iнформацiя має включати:
а) iнформацiю про вибiр та заплановану функцiю пристрою для введення та демонстрацiю сумiсностi пристрою для введення з iншими складовими продукту;
б) докази вiдповiдностi частини пристрою для введення загальним вимогам, встановленим Директивою 93/42/ЄЄС "Медичнi прилади. Медичне обладнання. Медичнi пристрої" або iншим чинним нормативним актом, вiдповiдностi частини активного медичного виробу, що iмплантується, загальним вимогам, встановленим Директивою 90/385/ЄЄС "Активнi медичнi пристрої, що iмплантують";
в) за необхiдностi доказ вiдповiдностi пристрою для введення або активного медичного виробу, що iмплантується, посилаючись на Керiвництво з мiнiмiзацiї ризику передачi збудникiв губчатої енцефалопатiї тварин з лiкарськими засобами (дiюче видання, опублiковане Комiсiєю в Офiцiйному бюлетенi Європейського союзу);
г) за наявностi/на вимогу Центру результати будь-якої оцiнки компетентним органом, що проводив таку оцiнку, частини пристрою для введення або частини активного медичного виробу, що iмплантується, проведеної компетентним органом на вiдповiднiсть вимогам Директиви 93/42/ЄЄС "Медичнi прилади. Медичне обладнання. Медичнi пристрої" або Директиви 90/385/ЄЄС "Активнi медичнi пристрої, що iмплантують".
12.3. Спецiальнi вимоги до Модуля 4:
Загальноприйнятi вимоги до матерiалiв реєстрацiйного досьє, викладених у Модулi 4 щодо доклiнiчного дослiдження лiкарських засобiв, не завжди можуть застосовуватись щодо матерiалiв реєстрацiйного досьє для лiкарських засобiв генної та соматоклiтинної терапiї у зв'язку з винятковими та рiзноманiтними структурними та бiологiчними властивостями таких лiкарських засобiв, включаючи високий ступiнь специфiчностi видiв, специфiчнiсть суб'єкта, iмунологiчнi бар'єри та вiдмiнностi у плейотропних реакцiях.
У Модулi 2 необхiдно надати вiдповiдне обґрунтування доклiнiчної розробки та критерiїв, що використанi для вибору вiдповiдних видiв i моделей.
За потреби необхiдно iдентифiкувати або розробити новi моделi на тваринах, щоб мати змогу екстраполювати на людину данi щодо специфiчної активностi та токсичностi. У матерiалах реєстрацiйного досьє необхiдно надати наукове обґрунтування використання цих моделей захворювання на тваринах для забезпечення безпеки та пiдтвердження ефективностi лiкарського засобу.
12.4. Спецiальнi вимоги до Модуля 5:
У матерiалах реєстрацiйного досьє необхiдно продемонструвати ефективнiсть високотехнологiчних лiкарських засобiв, як це описано у Модулi 5. Однак, може виявитися, що було неможливим проведення клiнiчних випробувань згiдно iз загальноприйнятими вимогами щодо певних лiкарських засобiв та певних терапевтичних показань. У матерiалах реєстрацiйного досьє Модуля 2 необхiдно надати обґрунтування будь-якого вiдхилення вiд iснуючих вимог.
У матерiалах реєстрацiйного досьє Модуля 5 повинно бути вiдображено тi особливостi, що може мати клiнiчна розробка лiкарських засобiв прогресивної терапiї через складний та лабiльний характер дiючих речовин. Це питання потребує додаткового розгляду через особливостi життєздатностi, пролiферацiї, мiграцiї та диференцiацiї клiтин (соматоклiтинна терапiя), через особливi клiнiчнi обставини, за яких використовуються лiкарськi засоби, або через особливий механiзм дiї, що здiйснюється через експресiю гена (генна терапiя).
У заявi про реєстрацiю високотехнологiчних лiкарських засобiв необхiдно зазначити особливий ризик, що пов'язаний з такими лiкарськими засобами та виникає через потенцiйне забруднення iнфекцiйними збудниками. Особливий акцент необхiдно зробити на раннiх стадiях розробки, включаючи вибiр донорiв, з одного боку, i терапевтичне втручання в цiлому, уключаючи вiдповiдне поводження з лiкарським засобом, та його застосування, з iншого боку.
Бiльше того, у вiдповiдних випадках матерiали реєстрацiйного досьє Модуля 5 повиннi мiстити iнформацiю про заходи щодо перевiрки та контролю функцiй i розвитку живих клiтин у реципiєнта для запобiгання передачi збудникiв iнфекцiї реципiєнту та мiнiмiзацiї потенцiйного ризику для здоров'я iнших людей, популяцiї.
12.4.1. Фармакологiчнi дослiдження та дослiдження ефективностi у людини:
Матерiали реєстрацiйного досьє щодо фармакологiчних дослiджень у людини повиннi надавати iнформацiю про очiкуваний механiзм дiї, очiкувану ефективнiсть, виходячи з виправданих кiнцевих цiлей, розподiлу в органiзмi, вiдповiдного режиму дозування та способiв введення або модальностi застосування, необхiдної для дослiдження бажаної ефективностi.
Проведення фармакокiнетичних дослiджень згiдно iз загальноприйнятими вимогами може виявитись неможливим для деяких високотехнологiчних лiкарських засобiв. Iнодi неможливо провести клiнiчнi дослiдження на здорових добровольцях, а визначення дози та кiнетики лiкарського засобу ускладнено. Однак у матерiалах реєстрацiйного досьє необхiдно надати iнформацiю про вивчення розподiлу та поведiнки лiкарського засобу in vivo, включаючи пролiферацiю клiтин та їх довгострокову функцiю, а також про ступiнь розподiлу генно-iнженерних лiкарських засобiв та тривалостi бажаної експресiї гена, про використання та, за необхiдностi, розробку вiдповiдних тестiв для монiторингу клiтинного препарату або клiтини, що експресує необхiдний ген в органiзмi людини, та для контролю функцiї клiтин, якi було введено або трансфектовано.
У матерiалах реєстрацiйного досьє має бути надана оцiнка ефективностi безпеки високотехнологiчного лiкарського засобу, що повинна включати докладний опис та оцiнку терапевтичної процедури в цiлому, включаючи особливi способи введення (такi як трансфекцiя клiтин ex vivo, манiпуляцiї in vitro або використання методiв iнтервенцiї) i випробування можливих додаткових режимiв лiкування (включаючи iмуносупресивне, антивiрусне та цитотоксичне).
Усю процедуру необхiдно апробувати при проведеннi клiнiчних випробувань та викласти в iнформацiї про лiкарський засiб.
12.4.2. Надання даних щодо безпеки:
У матерiалах реєстрацiйного досьє необхiдно вiдобразити питання безпеки, що виникають у зв'язку з iмунною реакцiєю на лiкарськi засоби або на експресованi бiлки i пов'язанi з iмунним вiдторгненням, iмунносупресiєю або вiдмовою iмуноiзоляцiйних структур.
Певнi лiкарськi засоби генної та соматоклiтинної терапiї (наприклад, ксеногеннi лiкарськi засоби та певнi лiкарськi засоби генного переносу) можуть мiстити реплiкацiйно-компетентнi частки та/або збудникiв iнфекцiй. Можливо на до- та пiсляреєстрацiйних стадiях необхiдно буде контролювати стан пацiєнта з точки зору розвитку можливих iнфекцiй та/або патологiчних ускладнень; можливо, такий контроль необхiдно розповсюдити на людей, з якими контактує пацiєнт, включаючи медичний персонал.
При використаннi деяких лiкарських засобiв соматоклiтинної терапiї та деяких лiкарських засобiв генного переносу неможливо повнiстю виключити ризик забруднення збудниками, передача яких потенцiйно можлива. Однак цей ризик можна мiнiмiзувати за допомогою вiдповiдних заходiв, описаних у матерiалах реєстрацiйного досьє Модуля 3.
Заходи, що є частиною виробничого процесу, повиннi доповнюватися супровiдними методами аналiзу, процедурами контролю якостi та вiдповiдними методами контролю, опис яких необхiдно включити до Модуля 5.
Застосування деяких лiкарських засобiв соматоклiтинної терапiї може обмежуватися тимчасово або постiйно закладами охорони здоров'я/установами, у яких є фахiвцi та обладнання для забезпечення специфiчного нагляду за безпекою пацiєнтiв. Подiбний пiдхiд може вiдповiдати певним лiкарським засобам генної терапiї, застосування яких пов'язане з потенцiйним ризиком наявностi збудникiв iнфекцiї, здатних до реплiкацiї.
У вiдповiдних випадках у матерiалах реєстрацiйного досьє необхiдно передбачити та висвiтлити питання тривалого нагляду за розвитком пiзнiх ускладнень.
У вiдповiдних випадках заявнику необхiдно надати детальний план процесу управлiння ризиками, що включає iнформацiю про клiнiчнi та лабораторнi данi пацiєнта, епiдемiологiчнi данi (якi виявляються) та, якщо це має вiдношення, архiвнi данi про зразки тканин вiд донора та реципiєнта. Така систематизована iнформацiя необхiдна для забезпечення монiторингу лiкарського засобу та швидкої реакцiї при виявленнi пiдозрiлих ознак небажаних явищ.
XI. ВИМОГИ ДО МАТЕРIАЛIВ РЕЄСТРАЦIЙНОГО ДОСЬЄ ДЛЯ АФI АБО ДIЮЧОЇ РЕЧОВИНИ
1. Вимоги до заяви:
У заявi має бути вказана назва АФI або дiючої речовини, технологiчна форма, включаючи упаковку.
Зазначаються найменування та мiсцезнаходження заявника разом з найменуванням та мiсцезнаходженням виробникiв i виробничих дiльниць, задiяних на рiзних стадiях виробництва, а також, за потреби, данi щодо iмпортера.
Сертифiкати якостi на три промисловi серiї АФI або дiючої речовини або сертифiкат якостi на одну промислову серiю у супроводi зобов'язання надати сертифiкати якостi на двi iншi серiї, як тiльки вони стануть доступними (усi сертифiкати якостi повиннi подаватися за кожним заявленим мiсцем виробництва).
2. Маркування упаковки:
Заявник надає пропонований текст маркування упаковки (первинної, вторинної (за наявностi)).
3. Оцiнка небезпеки для довкiлля:
У вiдповiдних випадках до заяви про державну реєстрацiю АФI або дiючої речовини додають резюме екологiчної оцiнки можливого ризику для довкiлля через її використання та/або утилiзацiю у складi лiкарського засобу, а також наводять пропозицiї щодо вiдповiдної iнформацiї, яка має бути наведена у маркуваннi. Необхiдно розглянути ризик для довкiлля, пов'язаний з вивiльненням АФI або дiючих речовин, що мiстять ГМО, вiдповiдно до вимог Закону України "Про державну систему бiобезпеки при створеннi, випробуваннi, транспортуваннi та використаннi генетично модифiкованих органiзмiв".
Iнформацiя повинна включати:
вступ;
копiю письмового дозволу, виданого уповноваженим органом країни виробника, на навмисне вивiльнення ГМО у навколишнє середовище при використаннi з дослiдницькою метою;
данi, включаючи методи виявлення та iдентифiкацiї, а також унiкальний код ГМО i будь-яку додаткову iнформацiю про ГМО або речовину, яка має значення при оцiнцi ризику для довкiлля;
звiт про оцiнку ризику для довкiлля (ОРД), пiдготовлений на пiдставi наявної iнформацiї;
вiдповiднi заходи для iнформування населення.
Наведена iнформацiя має бути засвiдчена пiдписом автора iз зазначенням дати, даних щодо освiти, стажування та професiйного досвiду. Необхiдно вказати професiйнi стосунки мiж автором i заявником.
4. Основнi данi:
4.1. Усi методики та методи, що використовуються при виробництвi та контролi АФI або дiючої речовини, слiд викладати чiтко i детально, щоб можна було вiдтворити їх при проведеннi контрольних випробувань, на вимогу уповноваженого органу. Усi методи випробувань повиннi вiдповiдати сучасному науковому рiвню, а також бути валiдованими. Слiд надавати результати дослiджень з валiдацiї. Якщо використовуються методи випробувань, включенi до ДФУ або Європейської фармакопеї, то цей виклад необхiдно замiнити вiдповiдним посиланням на монографiю(ї) та загальний(i) роздiл(и).
4.2. Для всiх речовин, вказаних у монографiях ДФУ або Європейської фармакопеї, необхiдно робити посилання на фармакопеї. Стосовно речовин, якi не вказано в цих фармакопеях, мають надаватися посилання на iншi фармакопеї, стандарти яких є не нижчими за стандарти ДФУ або Європейської фармакопеї.
Якщо методи аналiзу включено до ДФУ або Європейської фармакопеї, то немає необхiдностi наводити їх повний виклад, достатньо в кожному роздiлi, в якому планувався виклад цього методу, робити вiдповiдне посилання на монографiю та загальний роздiл.
4.3. Якщо вихiднi матерiали та сировина, дiючi або допомiжнi речовини не описано анi в ДФУ, анi в Європейськiй фармакопеї, анi в iнших фармакопеях, стандарти яких є не нижчими за стандарти ДФУ або Європейської фармакопеї, може бути прийнятним посилання на монографiю фармакопеї iншої країни. У таких випадках заявник повинен надати результати дослiджень з валiдацiї аналiтичних методик, описаних у монографiї, а також, за необхiдностi, переклад.
4.4. Якщо АФI або дiюча речовина i вихiдний матерiал описано в монографiї Європейської фармакопеї, заявник може надати сертифiкат вiдповiдностi Європейськiй фармакопеї, виданий Європейським директоратом з питань якостi лiкарських засобiв. Виробник речовини повинен пiдтвердити, що виробничий процес не змiнювався з часу видачi цього сертифiката вiдповiдностi.
4.5. Необхiдно описати особливi запобiжнi заходи щодо передачi губчатої енцефалопатiї тварин (сировина, отримана вiд жуйних тварин): на кожнiй стадiї виробничого процесу заявник повинен продемонструвати вiдповiднiсть використаних матерiалiв, посилаючись на вiдповiднi керiвництва з мiнiмiзацiї ризику передачi збудникiв губчатої енцефалопатiї тварин з лiкарськими засобами. Пiдтвердити це можна, або надавши сертифiкат вiдповiдностi Європейськiй фармакопеї щодо губчатої енцефалопатiї, виданий Європейським директоратом з питань якостi лiкарських засобiв, або шляхом надання наукових даних для обґрунтування цiєї вiдповiдностi.
4.6. Слiд надати iнформацiю про оцiнку ризику потенцiйного зараження стороннiми агентами (незалежно вiд того, мають вони вiрусне походження чи нi) згiдно з вимогами, викладеними в спецiальних керiвництвах, а також у загальних монографiях та загальних роздiлах ДФУ або Європейської фармакопеї.
4.7. Особливу увагу придiляють викладенню таких роздiлiв матерiалiв реєстрацiйного досьє:
4.7.1. Загальна iнформацiя:
Має бути надана iнформацiя про назву АФI або дiючої речовини, включаючи рекомендовану МНН, за наявностi - фармакопейну назву, зазначену в ДФУ, Європейськiй фармакопеї, та хiмiчну назву.
Має бути надана структурна формула, включаючи вiдносну та абсолютну стереохiмiю, молекулярна формула та вiдносна молекулярна маса. Для високотехнологiчних (бiотехнологiчних) дiючих речовин за потреби необхiдно надати схематичну послiдовнiсть амiнокислот i вiдносну молекулярну масу.
4.7.2. Виробництво:
а) заявник має надати опис виробничого процесу АФI або дiючої речовини;
б) необхiдно перелiчити усi матерiали, необхiднi для виробництва, iз зазначенням стадiї виробництва, на якiй використовується кожний матерiал. Необхiдно надати iнформацiю про якiсть i контроль цих матерiалiв, а також iнформацiю, яка доводить, що всi матерiали вiдповiдають вимогам щодо їхнього запланованого використання.
Необхiдно перелiчити вихiднi матерiали (сировину), а також надати документи щодо їх якостi та методiв контролю (за вiдсутностi сертифiката вiдповiдностi Європейськiй фармакопеї, виданого Європейським директоратом з питань якостi лiкарських засобiв);
в) необхiдно представити вiдповiднi методи контролю та критерiї прийнятностi на кожнiй критичнiй стадiї виробництва, iнформацiю про якiсть i контроль промiжних продуктiв, а також про процес валiдацiї та/або його аналiз (за вiдсутностi сертифiката вiдповiдностi Європейськiй фармакопеї, виданого Європейським директоратом з питань якостi лiкарських засобiв);
г) якщо присутностi потенцiйно патогенних стороннiх агентiв уникнути неможливо, то вихiдна речовина може використовуватися тiльки в тих випадках, коли подальша обробка забезпечує їхнє видалення та/або iнактивацiю, що має доводитися в роздiлi, який стосується оцiнки вiрусної безпеки.
4.7.3. Опис характеристик:
Необхiдно надати данi щодо структури та iнших характеристик АФI або дiючої речовини.
Слiд надати пiдтвердження структури АФI або дiючої речовини, ґрунтуючись на результатах фiзико-хiмiчних, та/або iмунохiмiчних, та/або бiологiчних методiв, а також надати iнформацiю про домiшки.
4.7.4. Контроль:
Необхiдно надати докладну iнформацiю про специфiкацiї, якi використовуються для посерiйного контролю дiючої речовини, обґрунтування вибору цих специфiкацiй, методiв аналiзу та їх валiдацiї (за вiдсутностi сертифiката вiдповiдностi Європейськiй фармакопеї, виданого Європейським директоратом з питань якостi лiкарських засобiв).
4.7.5. Система упаковка/укупорка:
Необхiдно надати опис контейнера та системи упаковка/укупорка i специфiкацiї її компонентiв. За можливостi необхiдно використовувати пакувальнi засоби, якi вiдповiдають вимогам ДФУ або Європейської фармакопеї.
4.7.6. Стабiльнiсть:
За необхiдностi слiд надати короткi вiдомостi про тип проведених дослiджень, використанi протоколи i отриманi пiд час дослiджень результати.
За необхiдностi слiд надати оформленi у вiдповiдному форматi докладнi результати дослiдження стабiльностi, включаючи вiдомостi про аналiтичнi методики, якi використовуються для одержання даних, i валiдацiю цих методик.
XII. ВИМОГИ ДО ГОМЕОПАТИЧНИХ ЛIКАРСЬКИХ ЗАСОБIВ, МАТЕРIАЛИ РЕЄСТРАЦIЙНОГО ДОСЬЄ ЯКИХ МОЖУТЬ НЕ МIСТИТИ НАУКОВИХ КЛIНIЧНИХ ДАНИХ, ТА ОБСЯГ ДАНИХ, НЕОБХIДНИХ ДЛЯ ПIДТВЕРДЖЕННЯ ЇХНЬОЇ ЯКОСТI
1. Гомеопатичнi лiкарськi засоби, матерiали реєстрацiйного досьє яких можуть не мiстити доказiв їх терапевтичної ефективностi, повиннi вiдповiдати таким умовам:
лiкарськi засоби призначенi для перорального або зовнiшнього застосування;
на етикетцi лiкарського засобу або в будь-якiй iнформацiї, що його стосується, не повиннi наводитися конкретнi показання для застосування;
ступiнь розведення лiкарського засобу є достатнiм для того, аби гарантувати його безпеку; зокрема лiкарський засiб не може мiстити бiльше однiєї частини на 10000 маткового розчину або 1/100 вiд мiнiмальної дози, яка використовується в алопатiї щодо субстанцiй, наявнiсть яких в алопатичному лiкарському засобi потребує обов'язкового пред'явлення рецепта лiкаря.
2. Заява може охоплювати одну серiю лiкарського засобу, одержаного з однiєї й тiєї самої гомеопатичної сировини або видiв сировини. Для того, аби продемонструвати, зокрема, фармацевтичну якiсть лiкарських засобiв та їх однорiднiсть вiд серiї до серiї, матерiали реєстрацiйного досьє мають включати:
наведену у фармакопеї (ДФУ, або Європейська фармакопея, або в разi вiдсутностi - Нiмецька гомеопатична фармакопея (GHP), Гомеопатична фармакопея США (HPUS), Британська гомеопатична фармакопея (BHP), Гомеопатична фармакопея Швабе) наукову назву гомеопатичної сировини або видiв сировини та данi про рiзнi шляхи введення, лiкарськi форми та ступенi розведення, якi пiдлягають реєстрацiї;
досьє з описом способу одержання та контролю гомеопатичної сировини або видiв сировини та обґрунтування його/їх гомеопатичної природи на основi вiдповiдної бiблiографiї;
опис виробництва та контролю для кожної лiкарської форми та опис способу розведення i пiдсилення дiї лiкарського засобу;
копiю лiцензiї на виробництво (якщо згiдно iз законодавством країни виробника лiцензiя на виробництво iснує лише в електронному виглядi (наприклад, США), має бути надана роздрукiвка iз посиланням на вiдповiдний офiцiйний сайт, засвiдчена пiдписом/печаткою заявника) або iншого дозвiльного документа на виробництво заявленої лiкарської форми у країнi виробника;
засвiдчену копiю документа, що пiдтверджує вiдповiднiсть виробництва лiкарського засобу вимогам GMP, виданого Держлiкслужбою вiдповiдно до вимог наказу Мiнiстерства охорони здоров'я України вiд 27 грудня 2012 року N 1130 "Про затвердження Порядку проведення пiдтвердження вiдповiдностi умов виробництва лiкарських засобiв вимогам належної виробничої практики", зареєстрованого у Мiнiстерствi юстицiї України 21 сiчня 2013 року за N 133/22665, або гарантiйного листа заявника щодо надання такого документа протягом строку проведення спецiалiзованої експертизи;
копiю реєстрацiйного посвiдчення або iншого дозвiльного документа (Marketing Authorization, Certificate of a Pharmaceutical Product, Free Sale Certificate тощо), виданого уповноваженим органом країни власника реєстрацiйного посвiдчення (заявника) та/або виробника, або iншої країни, регуляторний орган якої керується високими стандартами якостi, що вiдповiдають стандартам, рекомендованим ВООЗ, на ринку якої розмiшено лiкарський засiб. Зазначити перелiк країн, де цей лiкарський засiб зареєстровано/перереєстровано;
пропозицiї до маркування на упаковцi;
iнструкцiю для медичного застосування у разi, якщо на зовнiшнiй упаковцi наведено неповну iнформацiю, передбачену роздiлом XVIII цього Порядку;
данi про стабiльнiсть лiкарського засобу.
XIII. ОКРЕМI ВИМОГИ ДО ТРАДИЦIЙНИХ ЛIКАРСЬКИХ ЗАСОБIВ РОСЛИННОГО ПОХОДЖЕННЯ ТА ДО ЇХ МАТЕРIАЛIВ РЕЄСТРАЦIЙНОГО ДОСЬЄ
1. Допускається спрощена процедура реєстрацiї традицiйних лiкарських засобiв рослинного походження, що вiдповiдають таким умовам:
а) показання для застосування мають вiдповiдати традицiйним лiкарським засобам рослинного походження, якi завдяки своєму складу призначенi для самостiйного використання без контролю з боку лiкаря;
б) лiкарськi засоби призначенi виключно для застосовування вiдповiдно до точно вказаного дозування та схеми застосування (у вказаних дозах та вiдповiдно до наведеного режиму прийому);
в) лiкарськi засоби призначенi для перорального, зовнiшнього або iнгаляцiйного застосування;
г) перiод традицiйного застосування має становити понад 30 рокiв у свiтi i понад 15 рокiв в Українi;
ґ) є достатня кiлькiсть даних про традицiйне використання лiкарського засобу, зокрема доведена безпека застосування у звичайних умовах, фармакологiчнi ефекти або ефективнiсть лiкарського засобу доведенi тривалим використанням i досвiдом.
Наявнiсть у складi лiкарського засобу рослинного походження вiтамiнiв та мiнералiв, безпеку яких пiдтверджено задокументованими доказами, не повинна бути перешкодою для реєстрацiї таких лiкарських засобiв, як традицiйних лiкарських засобiв рослинного походження, за умови, що дiя вiтамiнiв або мiнералiв є допомiжною або здатна пiдсилювати дiю активних iнгредiєнтiв рослинного походження щодо заявлених показань.
2. Комплект реєстрацiйних документiв на традицiйнi лiкарськi засоби, що подаються на державну реєстрацiю, повинен мiстити докладнi вiдомостi, викладенi у Модулях 1, 2 та 3 роздiлiв IX i X цього Порядку.
Крiм того, необхiдно надати:
а) копiю реєстрацiйного посвiдчення або iншого дозвiльного документа (Marketing Authorization, Certificate of a Pharmaceutical Product, Free Sale Certificate тощо), виданого уповноваженим органом країни власника реєстрацiйного посвiдчення (заявника) та/або виробника, або iншої країни, регуляторний орган якої керується високими стандартами до якостi, що вiдповiдають стандартам, рекомендованим ВООЗ, на ринку якої розмiшено лiкарський засiб. Зазначити перелiк країн, де даний лiкарський засiб зареєстровано/перереєстровано;
б) бiблiографiчнi або експертнi данi (лiтературний огляд або експертний звiт) вiдносно ефектiв даного або аналогiчного лiкарського засобу, що використовувався у медичнiй практицi протягом понад 30 рокiв (з них понад 15 рокiв в Українi).
Аналогiчним лiкарським засобом рослинного походження вважається лiкарський засiб, який мiстить таку саму дiючу речовину, незалежно вiд використовуваних допомiжних речовин, призначений для застосування за такими самими або подiбними показаннями, з такою самою силою дiї та способом застосування, таким самим або подiбним шляхом введення, що й лiкарський засiб, на який подається заява;
в) бiблiографiчний огляд, що описує безпеку застосування даного лiкарського засобу та супроводжується експертним звiтом, а на вимогу Центру - додатковi данi, необхiднi для оцiнки безпеки лiкарського засобу;
г) копiю лiцензiї на виробництво (якщо згiдно iз законодавством країни виробника лiцензiя на виробництво iснує лише в електронному виглядi (наприклад, США), має бути надана роздрукiвка iз посиланням на вiдповiдний офiцiйний сайт, засвiдчена пiдписом/печаткою заявника) або iншого дозвiльного документа на виробництво заявленої лiкарської форми у країнi виробника;
ґ) засвiдчену копiю документа, що пiдтверджує вiдповiднiсть виробництва лiкарського засобу вимогам GMP, виданого Держлiкслужбою вiдповiдно до вимог наказу Мiнiстерства охорони здоров'я України вiд 27 грудня 2012 року N 1130 "Про затвердження Порядку проведення пiдтвердження вiдповiдностi умов виробництва лiкарських засобiв вимогам належної виробничої практики", зареєстрованого у Мiнiстерствi юстицiї України 21 сiчня 2013 року за N 133/22665, або гарантiйного листа заявника щодо надання такого документа протягом строку проведення спецiалiзованої експертизи.
3. Вимога надання документа про медичне використання протягом 30 рокiв вважається виконаною у випадку вiдсутностi реєстрацiї лiкарського засобу. Лiкарський засiб також вважається таким, що вiдповiдає данiй вимозi, навiть, якщо кiлькiсть дiючих речовин лiкарського засобу була знижена протягом цього перiоду.
4. У реєстрацiї традицiйного лiкарського засобу рослинного походження за спрощеною процедурою може бути вiдмовлено у разi, якщо лiкарський засiб не вiдповiдає вищенаведеному, та:
а) не наведено кiлькiсний та якiсний склад компонентiв, або
б) показання до застосування не вiдповiдають традицiйним лiкарським засобам рослинного походження, якi завдяки своєму складу призначенi для самостiйного використання без контролю з боку лiкаря, або
в) препарат може бути небезпечним за звичайних умов застосування, або
г) данi, що пiдтверджують традицiйне застосування, недостатнi особливо, якщо фармакологiчнi ефекти або ефективнiсть не пiдтвердженi тривалим досвiдом застосування, або
ґ) не пiдтверджено фармацевтичну якiсть лiкарського засобу.
XIV. ОКРЕМI ВИМОГИ ДО ЛIКАРСЬКИХ ЗАСОБIВ, ЩО ВИРОБЛЯЮТЬСЯ ЗА ПРОПИСАМИ, ТА ДО ЇХ МАТЕРIАЛIВ РЕЄСТРАЦIЙНОГО ДОСЬЄ
1. Спрощена процедура реєстрацiї може бути застосована до певних лiкарських засобiв, що виробляються за прописами, якi вiдповiдають такому:
а) склад лiкарського засобу, технологiя виробництва, специфiкацiя та iнструкцiя для медичного застосування повиннi вiдповiдати таким, що наведенi у затверджених прописах;
б) дiючi речовини, застосованi у лiкарських засобах, мають вiдповiдати вимогам, наведеним у затверджених прописах;
в) до матерiалiв реєстрацiйного досьє допускається внесення тiльки таких змiн, якi зазначенi у цьому Порядку.
2. Комплект матерiалiв реєстрацiйного досьє на лiкарськi засоби, що виробляються згiдно iз затвердженими прописами, якi подаються на державну реєстрацiю, повинен мiстити докладнi вiдомостi, викладенi у Модулях 1 та 2 роздiлiв IX та X цього Порядку.
Крiм того, необхiдно надати:
а) методи контролю якостi та iнструкцiю для медичного застосування;
б) данi щодо дiючої речовини, якi включають найменування та мiсцезнаходження виробника, iнформацiю щодо виробництва, стабiльностi, маркування, пакування, умов зберiгання;
в) данi щодо готового лiкарського засобу, якi включають iнформацiю щодо об'єму серiй, стабiльностi, маркування, пакування, умов та термiну зберiгання;
г) копiю реєстрацiйного посвiдчення або iншого дозвiльного документа (Marketing Authorization, Certificate of a Pharmaceutical Product, Free Sale Certificate тощо), виданого уповноваженим органом країни власника реєстрацiйного посвiдчення (заявника) та/або виробника, або iншої країни, регуляторний орган якої керується високими стандартами якостi, що вiдповiдають стандартам, рекомендованим ВООЗ, на ринку якої розмiшено лiкарський засiб. Зазначити перелiк країн, де даний лiкарський засiб зареєстровано/перереєстровано;
ґ) копiю лiцензiї на виробництво (якщо згiдно iз законодавством країни виробника лiцензiя на виробництво iснує лише в електронному виглядi (наприклад, США), має бути надана роздрукiвка iз посиланням на вiдповiдний офiцiйний сайт, засвiдчена пiдписом/печаткою заявника) або iншого дозвiльного документа на виробництво заявленої лiкарської форми у країнi виробника;
д) засвiдчену копiю документа, що пiдтверджує вiдповiднiсть виробництва лiкарського засобу вимогам GMP, виданого Держлiкслужбою вiдповiдно до вимог наказу Мiнiстерства охорони здоров'я України вiд 27 грудня 2012 року N 1130 "Про затвердження Порядку проведення пiдтвердження вiдповiдностi умов виробництва лiкарських засобiв вимогам належної виробничої практики", зареєстрованого у Мiнiстерствi юстицiї України 21 сiчня 2013 року за N 133/22665, або гарантiйного листа заявника щодо надання такого документа протягом строку проведення спецiалiзованої експертизи;
е) лист-пiдтвердження щодо того, що склад, виробництво та контроль лiкарського засобу вiдповiдають пропису, включеному до Перелiку лiкарських засобiв, що виробляються згiдно iз затвердженими прописами, затвердженого наказом Мiнiстерства охорони здоров'я України вiд 11 червня 2009 року N 423.
XV. ОКРЕМI ВИМОГИ ДО ЛIКАРСЬКИХ ЗАСОБIВ ОБМЕЖЕНОГО ЗАСТОСУВАННЯ (ПРЕПАРАТIВ-СИРIТ) ТА ДО ЇХ МАТЕРIАЛIВ РЕЄСТРАЦIЙНОГО ДОСЬЄ
1. Лiкарськi засоби обмеженого застосування (препарати-сироти) повиннi вiдповiдати таким умовам:
а) призначенi для дiагностики, профiлактики або лiкування рiдкiсного захворювання, що загрожує життю або серйозно ослаблює iмунiтет не бiльше нiж 5 осiб на 10000 жителiв на дату подання заяви або призначенi для дiагностики, профiлактики або лiкування захворювання, що загрожує життю або серйозно ослаблює iмунiтет, або хронiчних захворювань, та їх продаж без надання компенсацiї вiд держави або спiвтовариства не принесе значного прибутку, який компенсує витрати на лiкарський засiб, понесенi виробником:
б) не iснує будь-якого iншого затвердженого задовiльного методу дiагностики, профiлактики чи лiкування даного захворювання або, за наявностi такого методу, заявлений лiкарський засiб принесе значну користь хворим.
2. Заява на реєстрацiю лiкарського засобу обмеженого застосування (препарату-сироти) подається вiдповiдно до додатка 1 до цього Порядку. До заяви додаються:
пiдтвердження внесення заявленого лiкарського засобу до реєстру Європейського спiвтовариства препаратiв-сирiт або
пiдтвердження статусу препарату-сироти, надане компетентними органами країни заявника/виробника або країн, у яких лiкарський засiб отримав цей статус, та вiдомостi про реєстрацiю його в iнших країнах як препарату-сироти;
письмове зобов'язання про надання щорiчного пiдтвердження статусу препарату-сироти, отримане вiд Комiтету по препаратах-сиротах Європейського спiвтовариства або компетентних органiв країни заявника/виробника або країн, у яких лiкарський засiб отримав цей статус.
3. Матерiали реєстрацiйного досьє оформлюються вiдповiдно до вимог роздiлiв IX та X цього Порядку (у форматi загального технiчного документа).
При поданнi матерiалiв реєстрацiйного досьє на лiкарськi засоби обмеженого застосування (препарати-сироти) можуть бути застосованi положення глави 5 роздiлу X цього Порядку. У такому випадку заявник у резюме доклiнiчних та клiнiчних даних повинен надати обґрунтування причин, за яких неможливо надати повну iнформацiю, та обґрунтування спiввiдношення користь/ризик для заявлених препаратiв-сирiт.
4. Заява повинна охоплювати тiльки тi терапевтичнi показання, якi вiдповiдають умовам, викладеним у пунктi 1 цього роздiлу. Це не може бути перешкодою для подання окремої заяви на нову реєстрацiю такого лiкарського засобу за iншими показаннями для застосування, що не вiдповiдають вищезазначеним умовам (за iншим реєстрацiйним досьє).
XVI. ВИМОГИ ДО IНСТРУКЦIЇ ДЛЯ МЕДИЧНОГО ЗАСТОСУВАННЯ ЛIКАРСЬКОГО ЗАСОБУ/ЛIКАРСЬКОГО ЗАСОБУ (МЕДИЧНОГО IМУНОБIОЛОГIЧНОГО ПРЕПАРАТУ)
1. Iнструкцiя для медичного застосування, що супроводжує лiкарський засiб, повинна бути складена вiдповiдно до короткої характеристики лiкарського засобу, структуру якої наведено у додатку 14 (у разi наявностi), або затверджена вiдповiдно до нормативних вимог країни заявника/виробника або країни, регуляторний орган якої керується високими стандартами якостi, що вiдповiдають стандартам, рекомендованим ВООЗ, та/або результатiв клiнiчних випробувань.
2. Iнструкцiя для медичного застосування супроводжує лiкарський засiб, якщо iнформацiя, що має мiститися в iнструкцiї, не наведена безпосередньо на вториннiй або первиннiй упаковцi лiкарського засобу.
3. Текст iнструкцiї для медичного застосування лiкарського засобу повинен бути викладений чiтко, українською мовою. За бажанням виробника/заявника, поряд iз текстом українською мовою додатково текст iнструкцiї може бути викладений регiональною мовою або мовою меншин за умови, що в текстах рiзними мовами буде наведено iдентичну iнформацiю.
4. Стосовно певних лiкарських засобiв може бути вiдмiнена вимога щодо необхiдностi вказувати в iнструкцiї для медичного застосування деякi данi про застосування лiкарського засобу за умови, що лiкарський засiб не призначений для самостiйного застосування пацiєнтом.
5. Розмiр шрифту повинен бути великим, наскiльки це можливо для зручностi, та читабельним. Мiнiмальнi вимоги включають: шрифт розмiром 8 пунктiв Дiдо з мiжрядковим iнтервалом не менше 1 мм.
6. У верхньому правому кутку iнструкцiї для медичного застосування, яка вкладається у вторинну упаковку лiкарського засобу, має бути зазначено номер та дата наказу МОЗ України про державну реєстрацiю/перереєстрацiю/внесення змiн у реєстрацiйнi матерiали лiкарського засобу, яким даний лiкарський засiб дозволено для медичного застосування в Українi, а також номер реєстрацiйного посвiдчення.
7. Iнструкцiя для медичного застосування повинна мiстити таку iнформацiю:
7.1. Назва лiкарського засобу.
7.2. Повний якiсний склад (стосовно дiючих та допомiжних речовин) i кiлькiсний склад (стосовно дiючих речовин) з використанням загальноприйнятих назв для кожної форми випуску лiкарського засобу (дозування та упаковка).
7.3. Лiкарська форма.
Основнi фiзико-хiмiчнi властивостi.
7.4. Фармакотерапевтична група. Код АТХ.
7.5. Фармакологiчнi властивостi та, якщо така iнформацiя є корисною для лiкування, фармакокiнетичнi характеристики або iмунологiчнi i бiологiчнi властивостi.
Роздiл "Фармакологiчнi властивостi" на лiкарськi засоби, що вiдпускаються без рецепта, повинен бути викладений зрозумiлою мовою для пацiєнта та, за бажанням заявника, iнформацiя може бути скорочена.
Роздiл "Iмунологiчнi i бiологiчнi властивостi" є роздiлом iнструкцiї для медичного застосування певних категорiй медичних iмунобiологiчних препаратiв (вакцин, анатоксинiв тощо).
7.6. Клiнiчнi характеристики:
Терапевтичнi показання.
7.7. Протипоказання.
7.8. Особливi заходи безпеки (у разi необхiдностi), яких потрiбно дотримуватися особам, якi працюють з лiкарськими засобами та вводять їх пацiєнтам, а також усi заходи безпеки, яких необхiдно дотримуватися пацiєнту.
7.9. Взаємодiя з iншими лiкарськими засобами та iншi види взаємодiй.
7.10. Особливостi застосування:
7.10.1. Перерахувати та надати iнформацiю про всi допомiжнi речовини, знання про наявнiсть яких є важливими для безпечного та ефективного застосування лiкарського засобу (згiдно iз додатком 16 до цього Порядку).
7.10.2. Застосування у перiод вагiтностi або годування груддю.
7.10.3. Здатнiсть впливати на швидкiсть реакцiї при керуваннi автотранспортом або iншими механiзмами.
7.11. Дози i спосiб введення для дорослих.
7.11.1. Дiти.
7.12. Передозування (симптоми, невiдкладнi заходи лiкування та антидоти).
7.13. Побiчнi реакцiї (частота та вираженiсть за умови наявностi).
7.14. Термiн придатностi (у разi необхiдностi вказується термiн зберiгання пiсля пiдготовки лiкарського засобу для безпосереднього застосування, наприклад пiсля розчинення або пiсля першого вiдкриття первинної упаковки).
7.15. Умови зберiгання.
7.16. Несумiснiсть (основнi види) за наявностi iнформацiї.
7.17. Тип i мiсткiсть первинної упаковки.
7.18. Особливi заходи безпеки при поводженнi з невикористаними лiкарськими засобами та вiдходами лiкарських засобiв у разi необхiдностi.
7.19. Категорiя вiдпуску лiкарського засобу, встановлена пiд час державної реєстрацiї/перереєстрацiї/внесення змiн до матерiалiв реєстрацiйного досьє з урахуванням вимог наказу Мiнiстерства охорони здоров'я України вiд 17 травня 2001 року N 185 "Про затвердження критерiїв визначення категорiй вiдпуску лiкарських засобiв", зареєстрованого у Мiнiстерствi юстицiї України 31 травня 2001 року за N 464/5655.
7.20. Найменування та мiсцезнаходження виробника та адреса його мiсця провадження дiяльностi (зазначається виробник, вiдповiдальний за випуск серiї лiкарського засобу), а за бажанням заявника - найменування i мiсцезнаходження заявника та/або представника заявника.
7.21. Дата останнього перегляду iнструкцiї для медичного застосування (зазначається дата наказу МОЗ про реєстрацiю/перереєстрацiю/внесення змiн до iнструкцiї для медичного застосування лiкарського засобу пiд час дiї реєстрацiйного посвiдчення).
7.22. Для радiофармацевтичних лiкарських засобiв - повний докладний опис внутрiшньої радiацiйної дозиметрiї, а також додатковi докладнi iнструкцiї щодо приготування для негайного застосування та контролю якостi таких лiкарських засобiв, а у разi необхiдностi - максимальний термiн зберiгання, протягом якого будь-який промiжний лiкарський засiб, такий, як елюат, або готовий для застосування фармацевтичний лiкарський засiб буде вiдповiдати власним специфiкацiям.
Крiм того, iнструкцiя для медичного застосування радiофармацевтичного лiкарського засобу повинна мiстити опис усiх заходiв безпеки, яких необхiдно дотримуватися медичним працiвникам та пацiєнтам пiд час приготування та застосування лiкарського засобу, а також спецiальнi заходи безпеки з утилiзацiї упаковки та її невикористаного вмiсту.
8. В iнструкцiї для медичного застосування можуть бути розмiщенi символи або пiктограми, якi пояснюють iнформацiю, надану в нiй, а також iнша iнформацiя, яка вiдповiдає короткiй характеристицi лiкарського засобу i є корисною для санiтарної просвiти, за винятком iнформацiї рекламного характеру, яка сприяє просуванню лiкарського засобу на ринку.
9. Iнструкцiя для медичного застосування традицiйного лiкарського засобу рослинного походження повинна мiстити вказiвки на те, що:
лiкарський засiб є звичайним лiкарським засобом рослинного походження для застосування вiдповiдно до показань, пiдтверджених тривалим застосуванням;
користувач повинен проконсультуватися з лiкарем, якщо симптоми захворювання не зникли пiд час застосування лiкарського засобу або спостерiгаються побiчнi реакцiї, не зазначенi в iнструкцiї для медичного застосування лiкарського засобу.
10. Iнструкцiя для медичного застосування гомеопатичних лiкарських засобiв, якi вiдповiдають вимогам, викладеним у роздiлi XII цього Порядку, повинна мiстити:
формулювання "гомеопатичний лiкарський засiб без затверджених терапевтичних показань для застосування";
застереження для споживача про необхiднiсть консультацiї з лiкарем, якщо симптоми зберiгаються при застосуваннi лiкарського засобу.
11. Власник реєстрацiйного посвiдчення повинен гарантувати, що у разi необхiдностi зможе надати органiзацiям пацiєнтiв з обмеженим зором текст iнструкцiї для медичного застосування у доступному для них форматi.
XVII. ВИМОГИ ДО КОРОТКОЇ ХАРАКТЕРИСТИКИ ЛIКАРСЬКОГО ЗАСОБУ/ЛIКАРСЬКОГО ЗАСОБУ (МЕДИЧНОГО IМУНОБIОЛОГIЧНОГО ПРЕПАРАТУ)
1. Коротка характеристика лiкарського засобу (Summary of Product Characteristics - SPC) - iнформацiя для спецiалiстiв щодо безпеки та ефективностi застосування лiкарського засобу, яка затверджується за бажанням заявника.
2. Текст короткої характеристики лiкарського засобу повинен бути викладений чiтко, українською мовою.
3. Коротка характеристика лiкарського засобу, структуру якої наведено у додатку 14 до цього Порядку, повинна мiстити таку iнформацiю:
3.1. Назва лiкарського засобу, до складу якої у разi необхiдностi може входити дозування та лiкарська форма.
3.2. Повний якiсний склад (стосовно дiючих та допомiжних речовин) i кiлькiсний склад стосовно дiючих речовин з використанням загальноприйнятих назв для кожної форми випуску лiкарського засобу (дозування та упаковка).
3.3. Лiкарська форма.
3.4. Клiнiчна iнформацiя:
3.4.1. Терапевтичнi показання.
3.4.2. Нозологiя i спосiб застосування для дорослих i, якщо необхiдно, для дiтей.
3.4.3. Протипоказання.
3.4.4. Особливi застереження та запобiжнi заходи при застосуваннi, а у випадку медичного iмунобiологiчного препарату - обов'язковi запобiжнi заходи, що повиннi бути вжитi лiкарями, якi працюють з цими засобами та вводять їх пацiєнтам, а також усi запобiжнi заходи, яких повинен дотримуватися пацiєнт.
3.4.5. Взаємодiя з iншими лiкарськими засобами та iншi види взаємодiй.
3.4.6. Застосування пiд час вагiтностi та годування груддю.
3.4.7. Вплив на здатнiсть керувати транспортними засобами або працювати з iншими автоматизованими системами.
3.4.8. Побiчнi реакцiї.
3.4.9. Передозування (симптоми, невiдкладнi заходи лiкування, антидоти).
3.5. Фармакологiчнi властивостi:
3.5.1. Фармакотерапевтична група. Код АТХ.
3.5.2. Фармакодинамiчнi властивостi.
3.5.3. Фармакокiнетичнi властивостi.
3.5.4. Доклiнiчнi данi з безпеки.
3.6. Фармацевтична iнформацiя:
3.6.1. Перелiк допомiжних речовин.
3.6.2. Основнi випадки несумiсностi.
3.6.3. Термiн придатностi, у разi необхiдностi вказується термiн придатностi пiсля розчинення лiкарського засобу або пiсля першого вiдкриття первинної упаковки.
3.6.4. Особливi запобiжнi заходи при зберiганнi.
3.6.5. Тип та вмiст первинної упаковки.
3.6.6. Спецiальнi заходи безпеки при поводженнi з невикористаним лiкарським засобом або вiдходами лiкарського засобу (у разi необхiдностi).
3.7. Власник реєстрацiйного посвiдчення.
Виробник лiкарського засобу (найменування та мiсцезнаходження виробника та адреса його мiсця провадження дiяльностi (зазначається виробник, вiдповiдальний за випуск серiї лiкарського засобу).
3.8. Номер(и) реєстрацiйного(их) посвiдчення(нь).
3.9. Дата першої реєстрацiї або перереєстрацiї лiкарського засобу.
3.10. Дата перегляду тексту короткої характеристики.
3.11. Для радiофармацевтичних лiкарських засобiв - повний докладний опис внутрiшньої радiацiйної дозиметрiї.
3.12. Для радiофармацевтичних лiкарських засобiв - додатковi вказiвки для приготування лiкарських засобiв для негайного застосування та контролю якостi такого лiкарського засобу, а у разi необхiдностi - максимальний термiн зберiгання, протягом якого будь-який промiжний лiкарський засiб, наприклад елюат, або готовий до застосування лiкарський засiб буде вiдповiдати власним специфiкацiям.
XVIII. ВИМОГИ ДО МАРКУВАННЯ НА УПАКОВЦI ЛIКАРСЬКОГО ЗАСОБУ
1. Iнформацiя, що наноситься на упаковку (етикетку).
На вториннiй упаковцi готового лiкарського засобу, а за її вiдсутностi - на первиннiй упаковцi вказуються такi загальнi вiдомостi:
назва лiкарського засобу;
найменування та мiсцезнаходження виробника;
номер реєстрацiйного посвiдчення;
номер серiї;
способи застосування;
доза дiючої речовини в кожнiй одиницi та їх кiлькiсть в упаковцi;
термiн придатностi;
умови зберiгання;
запобiжнi заходи.
2. Вимоги до маркування упаковки лiкарського засобу.
2.1. Вимоги до вiдомостей, що вказуються на упаковцi.
2.1.1. Вторинна упаковка лiкарського засобу, а за її вiдсутностi - первинна упаковка має мiстити такi вiдомостi:
а) штрих-код лiкарського засобу;
б) назву лiкарського засобу, яка супроводжується зазначенням МНН або у разi її вiдсутностi загальноприйнятої назви (коли лiкарський засiб мiстить лише одну дiючу речовину); якщо назва лiкарського засобу може застосовуватися у декiлькох лiкарських формах та/або мати рiзну силу дiї, то необхiдно вказати лiкарську форму та/або силу дiї iз зазначенням того, чи призначений цей лiкарський засiб для дiтей вiком до 1 року, старше 1 року або дорослих;
в) дiючi речовини у якiсному та кiлькiсному вираженнi iз зазначенням їхнього вмiсту в одиницi дози або, залежно вiд способу застосування, в одиницi об'єму чи маси, з використанням їх мiжнародних непатентованих або загальноприйнятих назв;
г) лiкарську форму iз зазначенням маси, об'єму або кiлькостi одиниць дозування, що мiстяться в упаковцi;
ґ) перелiк допомiжних речовин згiдно з додатком 16 до цього Порядку, стосовно яких вiдомо, що вони спричиняють певну дiю або ефект i їх назви зазначаються разом з формулюванням: "для детальної iнформацiї див. iнструкцiю для медичного застосування". Однак на упаковцi повиннi бути вказанi всi допомiжнi речовини, якщо лiкарський засiб використовується парентерально, або в офтальмологiчнiй практицi, або для мiсцевого застосування (нашкiрнi та трансдермальнi лiкарськi засоби, лiкарськi засоби для iнгаляцiй, для ротової порожнини, назальнi, ректальнi та вагiнальнi лiкарськi засоби, тобто такi, що мають мiсцеву або трансдермальну дiю);
д) спосiб, а за необхiдностi - шлях введення лiкарського засобу;
е) особливi застереження щодо того, чи слiд зберiгати лiкарський засiб у недоступному для дiтей мiсцi i, за необхiдностi - поза полем зору дiтей;
є) дату закiнчення термiну придатностi (мiсяць/рiк) (зазначається останнiй мiсяць, коли термiн придатностi дiйсний);
ж) за необхiдностi особливi вказiвки вiдносно того, що робити з невикористаним лiкарським засобом або вiдходами, якi залишаються пiсля використання такого лiкарського засобу, а також, за бажанням заявника, посилання на будь-яку придатну систему збирання вiдходiв на мiсцi;
з) найменування та мiсцезнаходження виробника та адресу його мiсця провадження дiяльностi (зазначається виробник, вiдповiдальний за випуск серiй лiкарського засобу) i за необхiдностi найменування та мiсцезнаходження заявника або представника заявника.
У разi здiйснення виробництва лiкарського засобу з пакування in bulk, поряд з найменуванням виробника зазначається: "виробництво з пакування in bulk";
и) номер реєстрацiйного посвiдчення;
i) номер серiї лiкарського засобу, присвоєний виробником;
ї) якщо лiкарський засiб призначено для самостiйного лiкування, iнформацiю для його застосування;
й) за необхiдностi особливi застереження стосовно лiкарського засобу;
к) умови зберiгання, а за необхiдностi - особливi умови зберiгання.
У разi вiдсутностi вiдповiдної площi на упаковцi для нанесення повної iнформацiї обов'язково наносяться данi, перелiченi в пiдпунктах "а", "б", "в", "г", "є", "з", "и", "i", "й", "к" цього пункту, за умови наявностi iнструкцiї для медичного застосування.
2.1.2. На вториннiй упаковцi можуть бути також вмiщенi символи або пiктограми, якi дають змогу пояснити iнформацiю, вказану в пiдпунктi 2.1.1 цього пункту, а також iнша iнформацiя, яка вiдповiдає короткiй характеристицi лiкарського засобу та є корисною для пацiєнта, за винятком будь-яких елементiв рекламного характеру, якi сприяють просуванню цього лiкарського засобу на ринку.
2.1.3. Iнформацiя, зазначена в пiдпунктах 2.1.1 та 2.1.2 цього пункту, вказується на всiх первинних упаковках, за винятком випадкiв, описаних у пiдпунктi 2.1.4 цього пункту.
2.1.4. На первиннiй упаковцi у формi блiстера, стрипа тощо та на первиннiй упаковцi невеликого розмiру (ампулi, тюбику-крапельницi, шприцi-тюбику тощо), що вкладається у вторинну упаковку, яка вiдповiдає вимогам, викладеним у пiдпунктах пунктах 2.1.1, 2.1.2 цього пункту, зазначається як мiнiмум така iнформацiя:
а) назва лiкарського засобу;
б) маса, об'єм, концентрацiя або кiлькiсть одиниць дiї лiкарського засобу;
в) номер серiї лiкарського засобу;
г) дата закiнчення термiну придатностi;
ґ) найменування виробника та за необхiдностi заявника.
У разi вiдсутностi вiдповiдного мiсця на первиннiй упаковцi для нанесення зазначеної iнформацiї обов'язково зазначаються данi, перелiченi у пiдпунктах "а", "б", "в" цього пiдпункту.
2.2. Вимоги до маркування упаковки лiкарських засобiв, що мiстять радiонуклiди:
2.2.1. Вторинна упаковка та первинна упаковка лiкарських засобiв, що мiстять радiонуклiди, повиннi бути маркованi вiдповiдно до правил безпечного транспортування радiоактивних матерiалiв та вiдповiдати таким вимогам:
2.2.2. Етикетка на захисному контейнерi повинна мiстити докладнi вiдомостi, вказанi в пiдпунктi 2.1.1 цього пункту. Додатково маркування на захисному контейнерi повинно повнiстю пояснювати кодування на флаконi та за необхiдностi мiстити зазначення кiлькостi одиниць радiоактивностi лiкарського засобу у дозi або цiлому флаконi iз зазначенням дати, а у разi необхiдностi часу, а також кiлькостi капсул або для рiдини - об'єм у мiлiлiтрах вмiсту флакона.
2.2.3. Маркування флакона повинно мiстити таку iнформацiю:
назву або код лiкарського засобу, включаючи назву або хiмiчний символ радiонуклiда;
номер серiї та дату закiнчення термiну придатностi;
мiжнародний символ радiоактивностi;
найменування виробника;
кiлькiсть одиниць радiоактивностi, як указано в пiдпунктi 2.2.2 цього пункту.
2.3. Вимоги до маркування упаковки гомеопатичних лiкарських засобiв:
Для гомеопатичних лiкарських засобiв, якi вiдповiдають вимогам роздiлу XII цього Порядку, на етикетцi упаковки має бути наведена тiльки така iнформацiя:
штрих-код лiкарського засобу;
наукова назва сировини або видiв сировини iз зазначенням ступеня розведення iз застосуванням символiв ДФУ або iншої фармакопеї (Європейська фармакопея, Нiмецька гомеопатична фармакопея (GHP), Гомеопатична фармакопея США (HPUS), Британська гомеопатична фармакопея (BHP), Гомеопатична фармакопея Швабе);
найменування та мiсцезнаходження виробника та заявника i, за необхiдностi, найменування представника заявника;
спосiб застосування та, за необхiдностi, шлях введення;
дата закiнчення термiну придатностi (мiсяць/рiк) (зазначається останнiй мiсяць, коли термiн придатностi дiйсний);
лiкарська форма;
вмiст упаковки для продажу;
за необхiдностi, особливi умови зберiгання;
за необхiдностi, особливi застереження щодо лiкарського засобу;
номер серiї лiкарського засобу, присвоєний виробником;
номер реєстрацiйного посвiдчення;
формулювання "гомеопатичний лiкарський засiб без затверджених терапевтичних показань для застосування";
застереження для споживача про необхiднiсть консультацiї з лiкарем, якщо симптоми захворювання не зникли пiд час застосування лiкарського засобу.
2.4. Вимоги до маркування упаковки традицiйних лiкарських засобiв рослинного походження:
Для традицiйних лiкарських засобiв рослинного походження етикетка повинна мiстити докладнi вiдомостi, вказанi в пiдпунктах 2.1.1 та 2.1.2 цього пункту. Додатково повиннi бути наведенi вказiвки про те, що:
лiкарський засiб є традицiйним лiкарським засобом рослинного походження для використання вiдповiдно до показань, пiдтверджених тривалим застосуванням;
користувач повинен проконсультуватися з лiкарем, якщо симптоми захворювання не зникли пiд час застосування лiкарського засобу або спостерiгаються побiчнi реакцiї, не вказанi в iнструкцiї для медичного застосування.
2.5. Вимоги до маркування упаковки лiкарських засобiв, що мiстять один чи декiлька наркотичних засобiв та/або психотропних речовин:
На упаковцi лiкарських засобiв, що мiстять один чи декiлька наркотичних засобiв та/або психотропних речовин, повиннi мiститись докладнi вiдомостi, вказанi в пiдпунктах 2.1.1 та 2.1.2 цього пункту. Додатково первинна упаковка повинна бути позначена подвiйною червоною смугою.
2.6. Вимоги до тексту упаковки лiкарського засобу:
2.6.1. Данi на упаковцi мають бути нанесенi шрифтом не менше 7 пунктiв Дiдо.
2.6.2. Текст маркування викладається українською мовою. За бажанням виробника/заявника разом iз текстом маркування українською мовою додатково текст маркування може бути викладений регiональною мовою або мовою меншин за умови, що в текстах рiзними мовами буде наведено iдентичну iнформацiю.
2.6.3. Допускається, за необхiдностi, нанесення на упаковку iнформацiї за допомогою стикера, зокрема для оригiнальних (iнновацiйних) лiкарських засобiв (строком на один рiк з дати їх державної реєстрацiї), препаратiв обмеженого застосування (препаратiв-сирiт) або, за необхiдностi, в iнших випадках.
Iнформацiя, що мiститься на стикерi, має вiдповiдати вимогам глав 1 та 2 цього роздiлу. Стикер затверджується у складi Методiв контролю якостi лiкарського засобу при його реєстрацiї/перереєстрацiї/внесеннi змiн у реєстрацiйнi матерiали.
2.6.4. На вториннiй упаковцi лiкарських засобiв (крiм АФI або дiючих речовин та лiкарських засобiв в упаковцi in bulk) також шрифтом Брайля зазначаються назва лiкарського засобу, доза дiючої речовини та лiкарська форма вiдповiдно до Порядку маркування лiкарських засобiв шрифтом Брайля, затвердженого наказом Мiнiстерства охорони здоров'я України вiд 25 серпня 2010 року N 722, зареєстрованого у Мiнiстерствi юстицiї України 05 листопада 2010 року за N 1044/18339.
2.7. Вимоги щодо допомiжних речовин, якi мають бути нанесенi на упаковку:
2.7.1. Назви допомiжних речовин на упаковцi повиннi вказуватись у такий спосiб:
допомiжнi речовини мають бути представленi за їхнiми МНН вiдповiдно до Європейської фармакопеї або за вiдсутностi таких - загальноприйнятими назвами;
назву допомiжної речовини має супроводжувати номер "Е", якщо такий є. На упаковцi може бути зазначений тiльки номер "Е" за умови, що повна назва (МНН, а за її вiдсутностi - загальноприйнята назва) та номер "Е" буде наведено в iнструкцiї для медичного застосування у роздiлi "Склад";
патентованi коригенти або ароматизатори мають бути наданi у загальноприйнятих термiнах (наприклад, "апельсиновий смак", "лимонний аромат"); мають бути зазначенi будь-якi вiдомi основнi компоненти або такi, що мають певну визнану дiю або реакцiю;
хiмiчно модифiкованi допомiжнi речовини мають бути вказанi у такий спосiб, щоб уникнути плутанини з немодифiкованими допомiжними речовинами (наприклад, крохмаль попередньо желатинiзований);
усi компоненти складних допомiжних речовин або сумiшей мають бути заявленi, перелiченi в описi загального складу iз зазначенням основних складових речовин (наприклад, фарба для друку, що мiстить x, y, z). Загальна назва складної допомiжної речовини або сумiшi може бути використана на упаковцi у випадку, якщо бiльш повна iнформацiя надається в iнструкцiї для медичного застосування. Будь-який компонент, здатний спричинити певну дiю або реакцiю, має бути вказаний на упаковцi.
Цi вимоги не стосуються маркування на упаковках АФI або дiючих речовин.
2.7.2. Перелiк допомiжних речовин, якi мають бути обов'язково зазначенi на упаковцi, та iнформацiя щодо впливу цих речовин залежно вiд шляху введення та вмiсту допомiжної речовини, що має бути наведена в iнструкцiї для медичного застосування лiкарського засобу, наведено в додатку 16 до цього Порядку.
XIX. КРИТЕРIЇ ПРОВЕДЕННЯ ДОДАТКОВИХ ВИПРОБУВАНЬ ЛIКАРСЬКИХ ЗАСОБIВ ПРИ ПРОВЕДЕННI ЕКСПЕРТИЗИ МАТЕРIАЛIВ РЕЄСТРАЦIЙНОГО ДОСЬЄ
1. Додатковi випробування щодо фармацевтичної розробки при проведеннi експертизи матерiалiв реєстрацiйного досьє на лiкарськi засоби, що подаються на державну реєстрацiю, а також експертизи матерiалiв про внесення змiн до матерiалiв реєстрацiйного досьє протягом дiї реєстрацiйного посвiдчення проводяться у випадках, коли за результатами експертизи матерiалiв реєстрацiйного досьє матерiалiв було встановлено, що:
вiдсутнi або недостатнi данi, що мають бути представленi вiдповiдно до вимог, передбачених частиною II реєстрацiйного досьє роздiлiв VII та VIII цього Порядку або Модулем 3 роздiлiв IX та X цього Порядку, для пiдтвердження того, що розроблена лiкарська форма та запропонований склад задовольняють мету, вказану в заявi на проведення експертизи матерiалiв реєстрацiйного досьє;
вiдсутнi або недостатнi данi, що мають бути представленi вiдповiдно до вимог, передбачених частиною II реєстрацiйного досьє роздiлiв VII та VIII цього Порядку або Модулем 3 роздiлiв IX та X цього Порядку, щодо фiзико-хiмiчних характеристик дiючої речовини, якi впливають на бiодоступнiсть;
вiдсутнi або недостатнi данi щодо дослiджень для пiдтвердження еквiвалентностi генеричного лiкарського засобу до референтного препарату на етапi фармацевтичної розробки, передбаченi вимогами, викладеними у главi 4 роздiлу XX цього Порядку.
2. Додатковi випробування щодо пiдтвердження якостi лiкарських засобiв при проведеннi експертизи матерiалiв реєстрацiйного досьє на лiкарськi засоби проводяться у випадку, коли за результатами експертизи таких матерiалiв було встановлено, що результати клiнiчного аудиту свiдчать про недотримання умов зберiгання дослiджуваного лiкарського засобу при проведеннi клiнiчних випробувань, або виявлено невiдповiднiсть номерiв серiй зразкiв дослiджуваного лiкарського засобу при проведеннi клiнiчних випробувань вiдповiдно до вимог Порядку проведення клiнiчних випробувань лiкарських засобiв та експертизи матерiалiв клiнiчних випробувань, затвердженого наказом Мiнiстерства охорони здоров'я України вiд 23 вересня 2009 року N 690, зареєстрованого у Мiнiстерствi юстицiї України 29 жовтня 2009 року за N 1010/17026 (у редакцiї наказу Мiнiстерства охорони здоров'я України вiд 12 липня 2012 року N 523).
Додатковi випробування щодо пiдтвердження якостi лiкарських засобiв при проведеннi експертизи матерiалiв реєстрацiйного досьє на лiкарськi засоби не проводяться у випадках, зазначених у пiдпунктi 5.2 пункту 5 роздiлу XX цього Порядку.
3. Додатковi випробування щодо доклiнiчних дослiджень при проведеннi експертизи матерiалiв реєстрацiйного досьє на лiкарськi засоби проводяться у випадку, коли за результатами експертизи таких матерiалiв було встановлено, що данi, представленi вiдповiдно до вимог, передбачених частиною III реєстрацiйного досьє роздiлiв VII та VIII цього Порядку або Модулем 4 роздiлiв IX та X цього Порядку, щодо фармакологiчних, фармакокiнетичних та токсикологiчних дослiджень, необґрунтовано вiдсутнi або недостатнi.
4. Додатковi випробування щодо ефективностi та/або безпеки лiкарського засобу при проведеннi експертизи матерiалiв реєстрацiйного досьє на лiкарськi засоби проводяться у випадку, коли за результатами експертизи звiтiв проведеного(их) клiнiчного(их) дослiдження(ь), наданих заявником, встановлено, що:
клiнiчнi данi, наданi вiдповiдно до вимог частини IV реєстрацiйного досьє роздiлiв VII та VIII цього Порядку або Модуля 5 роздiлiв IX та X цього Порядку, представленi не в повному обсязi або/та недостатньо обґрунтовують окремi показання для застосування або протипоказання, схеми призначення;
наявнi негативнi данi стосовно ефективностi або безпеки лiкарського засобу, представленi вiдповiдно до вимог, передбачених частиною IV реєстрацiйного досьє роздiлiв VII та VIII цього Порядку або Модулем 5 роздiлiв IX та X цього Порядку, i цi данi потребують додаткових дослiджень;
проведенi клiнiчнi випробування є неадекватними для пiдтвердження ефективностi та безпеки лiкарського засобу;
данi спричиняють сумнiв щодо достовiрностi результатiв проведених клiнiчних випробувань.
5. Додатковi випробування для комбiнованих лiкарських засобiв, що мiстять у своєму складi вiдомi дiючi речовини, якi ранiше не застосовувались у даному сполученнi з терапевтичною метою, проводяться, якщо токсикологiчнi, фармакологiчнi або клiнiчнi дослiдження, викладенi вiдповiдно до вимог частини III реєстрацiйного досьє роздiлiв VII та VIII цього Порядку або Модуля 4 роздiлiв IX та X цього Порядку, наданi не у повному обсязi.
Це не стосується випадкiв, коли комбiнованi лiкарськi засоби мiстять вiдомi дiючi речовини, якi ранiше використовувались у медичнiй практицi як окремi препарати у тiй самiй лiкарськiй формi, дозах та вiдповiдному спiввiдношеннi для досягнення тiєї самої терапевтичної мети. При цьому заявник повинен надати результати дослiджень, якi пiдтверджують вiдсутнiсть будь-якої взаємодiї мiж iнгредiєнтами лiкарського засобу, яка може вплинути на його ефективнiсть та безпеку як на стадiї виробництва, так i протягом термiну зберiгання комбiнованого лiкарського засобу.
6. Додатковi випробування щодо бiодоступностi та еквiвалентностi генеричних лiкарських засобiв при проведеннi експертизи матерiалiв реєстрацiйного досьє проводяться у випадку, коли:
данi представлено не в повному обсязi для пiдтвердження еквiвалентностi лiкарського засобу вiдповiдно до вимог, викладених у пунктi 4 роздiлу XX цього Порядку;
данi, передбаченi вимогами, викладеними у пунктi 4 роздiлу XX цього Порядку, є неадекватними для пiдтвердження еквiвалентностi генеричного лiкарського засобу;
iснує ризик, що можливi вiдмiнностi у бiодоступностi можуть призвести до терапевтичної нееквiвалентностi.
7. Додатковi клiнiчнi випробування МIБП (клiнiко-епiдемiологiчнi випробування вакцин та анатоксинiв) проводяться у випадку, коли за результатами експертизи звiтiв клiнiчних випробувань (клiнiко-епiдемiологiчних випробувань для вакцин та анатоксинiв), наданих заявником, встановлено хоча б один iз таких фактiв:
у матерiалах реєстрацiйного досьє недостатньо даних для пiдтвердження показникiв ефективностi МIБП;
данi клiнiчних випробувань (клiнiко-епiдемiологiчних випробувань для вакцин та анатоксинiв) представлено не в повному обсязi або/та недостатньо обґрунтовують окремi показання для застосування або протипоказання, схеми призначення;
наявнi данi стосовно ефективностi або безпеки МIБП потребують додаткових перевiрок;
данi викликають сумнiв щодо достовiрностi результатiв проведених клiнiчних випробувань.
XX. ПОРЯДОК ПРОВЕДЕННЯ ДОДАТКОВИХ ВИПРОБУВАНЬ ЛIКАРСЬКИХ ЗАСОБIВ
1. Випробування щодо фармацевтичної розробки, пiдтвердження якостi та вiдтворюваностi методiв контролю якостi при проведеннi експертизи матерiалiв реєстрацiйного досьє проводяться в уповноважених лабораторiях вiдповiдно до галузi акредитацiї за направленням Центру.
Додатковi випробування щодо фармацевтичної розробки лiкарського засобу проводяться вiдповiдно до вимог, зазначених у частинi II реєстрацiйного досьє роздiлiв VII та VIII цього Порядку або Модулi 3 роздiлiв IX та X цього Порядку.
2. Додатковi випробування щодо доклiнiчних дослiджень лiкарських засобiв при проведеннi експертизи матерiалiв реєстрацiйного досьє проводяться вiдповiдно до вимог Порядку проведення доклiнiчного вивчення лiкарських засобiв та експертизи матерiалiв доклiнiчного вивчення лiкарських засобiв, затвердженого наказом Мiнiстерства охорони здоров'я України вiд 14 грудня 2009 року N 944, зареєстрованого у Мiнiстерствi юстицiї України 19 сiчня 2010 року за N 53/17348.
3. Додатковi випробування щодо ефективностi та/або безпеки лiкарських засобiв при проведеннi експертизи матерiалiв реєстрацiйного досьє проводяться вiдповiдно до вимог Порядку проведення клiнiчних випробувань лiкарських засобiв та експертизи матерiалiв клiнiчних випробувань (у редакцiї наказу Мiнiстерства охорони здоров'я України вiд 12 липня 2012 року N 523), затвердженого наказом Мiнiстерства охорони здоров'я України вiд 23 вересня 2009 року N 690, зареєстрованого в Мiнiстерствi юстицiї України 29 жовтня 2009 року за N 1010/17026.
4. Додатковi випробування щодо бiодоступностi та еквiвалентностi генеричних лiкарських засобiв при проведеннi експертизи матерiалiв реєстрацiйного досьє проводяться згiдно з вимогами Порядку проведення клiнiчних випробувань лiкарських засобiв та експертизи матерiалiв клiнiчних випробувань (у редакцiї наказу Мiнiстерства охорони здоров'я України вiд 12 липня 2012 року N 523), затвердженого наказом Мiнiстерства охорони здоров'я України вiд 23 вересня 2009 року N 690, зареєстрованого в Мiнiстерствi юстицiї України 29 жовтня 2009 року за N 1010/17026 (у випадку дослiдження еквiвалентностi за участю людини), та вимогами цього роздiлу.
Додатковi випробування щодо бiодоступностi та еквiвалентностi проводяться у закладах охорони здоров'я, а також лабораторiях фармакокiнетики Центру або в iнших уповноважених лабораторiях за направленням Центру.
4.1. Загальнi положення:
4.1.1. Мета проведення додаткових дослiджень встановлення еквiвалентностi (взаємозамiнностi) полягає в доведеннi еквiвалентностi (взаємозамiнностi) бiофармацевтичної якостi мiж генеричним та референтним лiкарськими засобами.
4.1.2. Для доведення еквiвалентностi (взаємозамiнностi) застосовуються такi методи:
порiвняльнi фармакокiнетичнi дослiдження за участю людини, у яких дiюча речовина та/або її метаболiти вимiрюються як функцiя часу у доступнiй бiологiчнiй рiдинi, а саме: кровi, плазмi, сироватцi або сечi для отримання фармакокiнетичних показникiв типу AUC i Cмакс, що вiдображають системну дiю;
порiвняльнi фармакодинамiчнi дослiдження за участю людини;
порiвняльнi клiнiчнi дослiдження;
дослiдження еквiвалентностi in vitro (процедура бiовейвер).
4.1.3. Прийнятнiсть кожного iз зазначених методiв для доведення еквiвалентностi (взаємозамiнностi) визначається на пiдставi комплексної експертизи факторiв, якi включають характеристики дiючої речовини, лiкарського засобу та часу дiї активної(их) речовини(н).
4.1.4. При виборi методiв доведення еквiвалентностi (взаємозамiнностi) застосовують такi пiдходи:
якщо можливо отримати вимiрюванi концентрацiї дiючої речовини в наявнiй бiологiчнiй рiдинi, такiй як плазма, проводяться порiвняльнi фармакокiнетичнi дослiдження за участю людини;
якщо неможливо отримати вимiрюванi концентрацiї дiючої речовини у вiдповiднiй бiологiчнiй рiдинi, проводяться порiвняльнi фармакодинамiчнi дослiдження за участю людини;
якщо неможливо визначити фармакокiнетичний профiль, знайти прийнятнi фармакодинамiчнi кiнцевi точки, проводяться порiвняльнi клiнiчнi дослiдження;
для лiкарських засобiв для орального застосування з негайним вивiльненням може застосовуватися дослiдження еквiвалентностi in vitro за процедурою бiовейвер вiдповiдно до БСК.
4.2. Вiдсутнiсть необхiдностi проведення дослiдження еквiвалентностi in vivo:
4.2.1. Генеричнi лiкарськi засоби, якi є фармацевтично еквiвалентними до референтних, вважаються терапевтично еквiвалентними (взаємозамiнними) i не потребують подальшого пiдтвердження еквiвалентностi шляхом проведення порiвняльних дослiджень при дотриманнi вимог до виробництва та стандартiв якостi, встановлених чинним законодавством України та мiжнародними нормами, за умов, що:
а) лiкарськi засоби для парентерального застосування (наприклад, внутрiшньовенно, пiдшкiрно або внутрiшньом'язово) у виглядi водних розчинiв, якi мiстять таку саму дiючу речовину i в тiй самiй молярнiй концентрацiї, що i референтний препарат, i з такими самими або подiбними допомiжними речовинами у порiвнюваних з референтним препаратом концентрацiях. Деякi допомiжнi речовини (зокрема буфернi розчини, консерванти, антиоксиданти) можуть вiдрiзнятися за умови доведення у будь-який спосiб, що у даних концентрацiях вони не впливають на безпеку та/або ефективнiсть лiкарського засобу;
б) лiкарськi засоби для орального застосування у формi розчинiв (наприклад, сиропи, елiксири i настойки), якi мiстять таку саму дiючу речовину в тiй самiй молярнiй концентрацiї, що й референтний препарат, i мiстять, в основному, такi самi допомiжнi речовини в порiвнюваних концентрацiях. Необхiдно ретельно вивчити допомiжнi речовини, про якi вiдомо, що вони впливають на проходження шлунково-кишковим трактом (далi - ШКТ), проникнiсть у ШКТ i, таким чином, на абсорбцiю та/або стабiльнiсть дiючої речовини у ШКТ;
в) лiкарськi засоби у формi порошкiв для приготування розчинiв, якщо розчин вiдповiдає вимогам пiдпунктiв "а" або "б" цього пункту;
г) лiкарськi засоби, якi є газами;
ґ) вушнi або очнi лiкарськi засоби, виготовленi у виглядi водних розчинiв, якi мiстять таку саму дiючу речовину(и) в такiй самiй молярнiй концентрацiї(ях) i, по сутi, такi самi допомiжнi речовини у порiвнюваних концентрацiях, що й референтний препарат. Деякi допомiжнi речовини (зокрема буфернi розчини, консерванти, речовини, якi коригують густину, або згущувачi) можуть вiдрiзнятися за умови доведення у будь-який спосiб, що у даних концентрацiях вони не впливають на безпеку та/або ефективнiсть лiкарського засобу;
д) лiкарськi засоби мiсцевої дiї у виглядi водних розчинiв, якi мiстять таку саму дiючу речовину(и) в такiй самiй молярнiй концентрацiї(ях) i, по сутi, такi самi допомiжнi речовини у порiвнюваних концентрацiях, що й референтний препарат;
е) лiкарськi засоби, якi є водними розчинами у формi iнгаляцiйно-розпилюваних небулайзером препаратiв, або назальнi спреї, що застосовуються за допомогою практично однакових пристроїв i якi мiстять таку саму дiючу речовину(и) в такiй самiй молярнiй концентрацiї(ях) i, по сутi, такi самi допомiжнi речовини у порiвнюваних концентрацiях, що й референтний препарат. Деякi допомiжнi речовини можуть вiдрiзнятися за умови доведення у будь-який спосiб, що у даних концентрацiях вони не впливають на безпеку та/або ефективнiсть лiкарського засобу;
є) лiкарськi засоби системної дiї у виглядi водних розчинiв для ректального або вагiнального застосування, якi мiстять таку саму дiючу(i) речовину(и) в такiй самiй молярнiй концентрацiї i, по сутi, такi самi допомiжнi речовини у порiвнюваних концентрацiях, що й референтний препарат. Деякi допомiжнi речовини можуть вiдрiзнятися за умови доведення у будь-який спосiб, що у даних концентрацiях вони не впливають на безпеку та/або ефективнiсть лiкарського засобу.
4.2.2. Для випадкiв, наведених у пiдпунктах "б", "в", "ґ", "д", "е" та "є" пiдпункту 4.2.1 цього пункту, у разi вiдмiнностей у складi допомiжних речовин заявник має довести, що при їх використаннi не передбачається вплив на безпеку та/або ефективнiсть лiкарського засобу.
У випадку, коли заявник не може надати таку iнформацiю i не має доступу до вiдповiдних даних, вiн повинен провести вiдповiднi дослiдження для доказу вiдсутностi впливу рiзних допомiжних речовин або допомiжних пристроїв на безпеку та/або ефективнiсть лiкарського засобу.
4.3. Необхiднiсть проведення дослiдження еквiвалентностi in vivo:
Доведення еквiвалентностi in vivo потрiбне у випадку, коли iснує ризик того, що можливi вiдмiнностi у бiодоступностi можуть призвести до терапевтичної нееквiвалентностi генеричного лiкарського засобу до референтного, наприклад, для:
а) лiкарських засобiв системної дiї для орального застосування з негайним вивiльненням, якщо до них застосованi один або декiлька таких критерiїв:
застосування лiкарського засобу для невiдкладної допомоги;
вузький спектр терапевтичної дiї (межа ефективнiсть/безпека), крутий нахил кривої доза - вiдклик;
документально пiдтвердженi проблеми щодо бiодоступностi або бiонееквiвалентностi, якi пов'язанi з дiючою речовиною або її формами (що не мають вiдношення до розчинення);
данi стосовно того, що на бiоеквiвалентнiсть можуть впливати полiморфiзм дiючої речовини, допомiжнi речовини та/або технологiчнi процеси;
б) лiкарських засобiв системної дiї, що не призначенi для орального або парентерального застосування (таких, як трансдермальнi пластирi, супозиторiї, нiкотиновi жувальнi гумки, гелi тестостерону та трансдермальнi контрацептиви);
в) лiкарських засобiв системної дiї з модифiкованим вивiльненням;
г) лiкарських засобiв системної дiї з фiксованою комбiнацiєю, у яких як мiнiмум для однiєї дiючої речовини потрiбно проведення дослiдження in vivo;
ґ) лiкарських засобiв несистемної дiї, зокрема для орального, назального, офтальмологiчного, дерматологiчного, ректального або вагiнального застосування, без системної абсорбцiї, що не є розчинами. У такому разi еквiвалентнiсть доводять шляхом проведення, наприклад, порiвняльних клiнiчних або фармакодинамiчних, дерматофармакокiнетичних дослiджень та/або дослiджень in vitro;
д) лiкарських засобiв, що мiстять вiдмiннi вiд референтного: ефiр, складний ефiр, iзомер, рацемат, комплекси або продукти дериватизацiї дiючої речовини, оскiльки цi вiдмiнностi можуть спричинити рiзну бiодоступнiсть.
4.4. Умови проведення процедури бiовейвер:
Застосування процедури бiовейвер для доведення еквiвалентностi генеричних лiкарських засобiв обмежується швидкорозчинними дiючими речовинами з вiдомою абсорбцiєю у людини, якi не мають вузького терапевтичного iндексу та критичного застосування.
Бiовейвер може застосовуватися для лiкарських засобiв у твердих дозованих формах системної дiї з негайним вивiльненням для орального застосування. Процедура бiовейвер не застосовується для сублiнгвальних, букальних лiкарських засобiв i препаратiв з модифiкованим вивiльненням. Для лiкарських засобiв, диспергованих у ротовiй порожнинi, пiдхiд бiовейвера на пiдставi БСК може застосовуватися лише тодi, коли абсорбцiя в ротовiй порожнинi може бути виключена.
4.4.1. Загальнi вимоги:
Бiовейвер на пiдставi БСК застосовується до лiкарських засобiв з негайним вивiльненням, якщо:
було доведено, що дiюча речовина проявляє високу розчиннiсть i повну абсорбцiю/високий ступiнь проникнення (I клас за БСК);
лiкарський засiб належить до дуже швидкорозчинних (> 85 % дiючої речовини переходить у розчин за 15 хвилин) або швидкорозчинних (85 % за 30 хвилин);
допомiжнi речовини, якi можуть вплинути на бiодоступнiсть, є якiсно i кiлькiсно подiбними. В основному використання подiбних допомiжних речовин у схожих кiлькостях є переважним
або:
було доведено, що дiюча речовина проявляє високу розчиннiсть i обмежену абсорбцiю/низький ступiнь проникнення (III клас за БСК);
лiкарський засiб належить до дуже швидкорозчинних (> 85 % дiючої речовини переходить у розчин за 15 хвилин);
допомiжнi речовини, якi можуть вплинути на бiодоступнiсть, є якiсно i кiлькiсно подiбними, й iншi допомiжнi речовини є якiсно подiбними i кiлькiсно дуже схожими.
В основному ризики невiдповiдного рiшення щодо можливостi проведення процедури бiовейвер мають бути критичнiше розглянутi для лiкарських засобiв, дiюча речовина яких вiдноситься до III класу за БСК (наприклад, абсорбцiя, специфiчна для певної дiлянки, ризик взаємодiї транспортних бiлкiв у дiлянцi абсорбцiї, склад допомiжних речовин i терапевтичнi ризики).
4.4.2. Дiюча речовина:
Процедура бiовейвер застосовується тiльки у випадках, коли дiюча речовина у дослiджуваному та референтному лiкарських засобах є тiєю самою. У випадку використання дiючої речовини у виглядi рiзних солей процедура бiовейвер можлива, якщо дiюча речовина вiдноситься до I класу за БСК.
Бiовейвер не застосовується, якщо дослiджуваний i референтний лiкарськi засоби мiстять рiзни ефiри, складнi ефiри, iзомери, рацемати, комплекси або продукти дериватизацiї дiючої речовини, оскiльки означенi вiдмiнностi можуть призвести до рiзної бiодоступностi, яка не може бути встановлена на пiдставi дослiджень на основi БСК та результатiв порiвняльних дослiджень in vitro з використанням тесту "Розчинення".
4.4.3. Розчиннiсть:
Необхiдно визначити та розглянути профiль pH-залежної розчинностi дiючої речовини. Дiюча речовина вважається високорозчинною, якщо найвища одноразова доза повнiстю розчиняється в 250 мл або в меншому об'ємi кожного з трьох буферних розчинiв iз значенням pH в межах 1 - 6,8. Встановлення рiвноважної розчинностi необхiдно проводити у трьох буферних розчинах при рекомендованих значеннях pH 1,2; 4,5 та 6,8, температурi (37±1) °C, та додатково враховуючи pKa, якщо її величина знаходиться у вищезазначеному дiапазонi pH, де pKa - константа iонiзацiї дiючої речовини. Повторнi дослiдження при кожному значеннi pH можуть бути необхiдними для досягнення чiткої класифiкацiї розчинностi дiючої речовини (наприклад, методом струшування колби або iншим обґрунтованим методом). Рекомендується вивчення розчинностi дiючої речовини проводити в трьох повторах при кожному iз вибраних значень pH для кожного заявленого виробника дiючої речовини. Значення pH кожного буферного розчину необхiдно перевiряти до та пiсля експерименту.
4.4.4. Всмоктування/абсорбцiя:
Демонстрацiя повної абсорбцiї у людини є переважною для проведення процедури бiовейвер. З цiєю метою вважається, що повна абсорбцiя має бути встановлена, якщо вимiрюваний рiвень абсорбцiї складає бiльше 85 %. Повна абсорбцiя в основному пов'язана з високою проникнiстю. Повна абсорбцiя препарату має бути обґрунтована на пiдставi дослiджень у людини.
Для вивчення ступеня абсорбцiї можуть використовуватися данi дослiджень абсолютної бiодоступностi або визначення масобалансу.
Якщо данi з встановлення масобалансу використовуються для пiдтвердження повної абсорбцiї, повинна бути гарантiя, що метаболiти, якi враховуються при визначеннi абсорбованої фракцiї, формуються пiсля абсорбцiї.
Дослiдження проникностi in vitro (in vivo або in situ кишкова перфузiя з використанням моделей тварин; in vitro проникнення крiзь iзольованi тканини тварин або моношар культури епiтелiальних клiтин) можуть також вважатися пiдтверджуючими для даних in vivo.
4.4.5. Готовий лiкарський засiб:
Вивчення кiнетики вивiльнення дiючої речовини генеричного та референтного лiкарських засобiв проводять у порiвняннi з референтним препаратом за таких умов:
прилади - прилад iз кошиком (100 об/хв) або прилад iз лопаттю (50 об/хв);
об'єм середовища розчинення - 900 мл або менше, але не менше 500 мл;
температура середовища розчинення - (37±0,5) °C;
кiлькiсть дослiджуваних зразкiв - 12 для дослiджуваного лiкарського засобу i 12 для референтного лiкарського засобу;
точки контролю - не менше 3 точок (не враховуючи нульову), а саме: через 10 хв., 15 хв., 20 хв., 30 хв. та 45 хв., причому точка 15 хв. є визначальною;
вiдносне стандартне вiдхилення середнього результату або коефiцiєнт варiацiї будь-якого лiкарського засобу повинен бути менше 20 % для першої точки i менше 10 % вiд другої до останньої точки контролю;
середовища розчинення (буфернi розчини): pH 1,0 - 1,2 (зазвичай 0,1N HCl або штучний шлунковий сiк без ферментiв), pH 4,5 i pH 6,8 (або штучна кишкова рiдина без ферментiв); (pH повинен гарантуватися впродовж всього дослiдження; рекомендуються буфернi розчини, описанi у Державнiй фармакопеї України або Європейськiй фармакопеї); iншi умови: вiдсутнiсть поверхнево-активних речовин; у випадку желатинових капсул або таблеток з желатиновим покриттям може допускатися використання ферментiв.
Необхiдно надати результати порiвняльних дослiджень in vitro, що включають протокол випробовування та умови їх проведення, iндивiдуальнi i середнi результати та вiдповiдну пiдсумкову статистику.
4.4.6. Оцiнка результатiв розчинення in vitro:
Лiкарськi засоби вважаються дуже швидкорозчинними, якщо не менше 85 % вiд зазначеної у маркуваннi кiлькостi дiючої речовини переходить у розчин за 15 хвилин або менше. У тому випадку, якщо це доведено для генеричного i референтного лiкарських засобiв, їх подiбнiсть може бути прийнята без будь-якого математичного обчислення.
Лiкарськi засоби вважаються швидкорозчинними, якщо не менше 85 % вiд зазначеної у маркуваннi кiлькостi дiючої речовини переходить у розчин за 30 хвилин.
Вiдсутнiсть вiдповiдних вiдмiнностей (схожiсть) має бути продемонстрована у випадках, якщо потрiбно бiльше 15 хвилин, але не бiльше 30 хвилин для досягнення майже повного розчинення (принаймнi 85 % вiд зазначеної у маркуваннi кiлькостi дiючої речовини). Фактор подiбностi або iншi вiдповiднi тести повиннi використовуватися для демонстрацiї профiлю подiбностi дослiджуваного та референтного лiкарських засобiв.
4.4.7. Допомiжнi речовини:
Для лiкарських засобiв I класу за БСК рекомендується використовувати схожу кiлькiсть тих самих допомiжних речовин у складi генеричного лiкарського засобу, як i у складi референтного лiкарського засобу.
Якщо бiовейвер застосовується до дiючих речовин III класу за БСК, допомiжнi речовини дослiджуваного та референтного лiкарських засобiв мають бути якiсно однаковими i кiлькiсно дуже схожими, щоб виключити рiзнi ефекти на мембраннi бiлки-транспортери.
Допомiжнi речовини, якi можуть впливати на бiодоступнiсть (наприклад, сорбiтол, манiтол, натрiю додецилсульфат або iншi поверхнево-активнi речовини), мають бути якiсно i кiлькiсно однаковими у дослiджуваному та референтному лiкарських засобах.
4.5. Загальнi аспекти проведення тесту "Розчинення":
Повинно бути достатньо точок вiдбору зразкiв для отримання значущих профiлiв розчинення, принаймнi через кожнi 15 хвилин. Рекомендується проводити частiший вiдбiр зразкiв пiд час перiоду найбiльшої змiни в профiлi розчинення. Для швидкорозчинних лiкарських засобiв, якщо повне розчинення вiдбувається протягом 30 хвилин, може бути необхiдний вiдповiдний профiль шляхом вiдбору зразкiв через кожнi 5 - 10 хвилин.
Якщо бiльше нiж 85 % лiкарського засобу розчиняється протягом 15 хвилин, профiлi розчинення можуть прийматися як подiбнi без подальшої математичної оцiнки.
У разi якщо не менше 85 % дiючої речовини не переходить у розчин за 15 хвилин, а переходить у розчин за 30 хвилин, то потрiбнi принаймнi три точки вiдбору зразкiв: перша точка вiдбору - до 15 хвилин, друга - 15 хвилин i третя, - коли вивiльнення досягає близько 85 %.
Для лiкарських засобiв з модифiкованим вивiльненням необхiдно дотримуватися таких вимог:
профiлi розчинення лiкарських форм з вiдтермiнованим вивiльненням мають бути подiбними в точках контролю, що вiдповiдають механiзму їх розчинення, тобто при pH = 1,2 (протягом 2 год.) i при pH = 6,8;
профiлi розчинення форм з пролонгованим вивiльненням мають бути подiбними у трьох середовищах розчинення у точках контролю, якi вiдповiдають швидкостi їх розчинення, наприклад, через 1 год., 2 год., 4 год., 6 год., 8 год. i 16 год.
Фактор подiбностi f2 обчислюють за формулою:
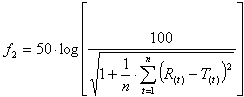 |
, |
де f2 - фактор подiбностi,
n - кiлькiсть точок контролю,
R(t) - середнє значення кiлькостi дiючої речовини, що перейшла у розчин при кожнiй зазначенiй точцi контролю референтного препарату (у вiдсотках до значення, вказаного на упаковцi),
T(t) - середнє значення кiлькостi дiючої речовини, що перейшла у розчин при кожнiй зазначенiй точцi контролю дослiджуваного препарату (у вiдсотках до значення, вказаного на упаковцi).
Для референтного та генеричного лiкарських засобiв має визначатися кiлькiсть дiючої речовини у вiдсотках вiд вказаного на етикетцi.
Оцiнка фактора подiбностi ґрунтується на таких умовах:
не менше трьох точок контролю (не враховуючи нульову);
точки контролю для дослiджуваного i референтного лiкарських засобiв повиннi збiгатися;
кiлькiсть дослiджуваних зразкiв - 12 повторюваностей для дослiджуваного лiкарського засобу i 12 - для референтного;
пiсля розчинення 85 % дiючої речовини з референтного лiкарського засобу до уваги беруться значення тiльки в однiй точцi контролю;
вiдносне стандартне вiдхилення середнього значення для кожного з лiкарських засобiв має бути менше 20 % у першiй точцi контролю i не бiльше 10 %, починаючи з другої i до останньої точки контролю.
Профiлi розчинення вважаються подiбними, якщо фактор подiбностi f2 і 50.
Необхiдно надати задокументованi результати порiвняльних дослiджень in vitro, включаючи протокол випробовування, iнформацiю про дослiджувану i референтну серiї лiкарських засобiв, докладнi данi щодо умов проведених випробувань, iндивiдуальнi i середнi результати та вiдповiдну пiдсумкову статистику.
5. Додатковi випробування щодо пiдтвердження якостi лiкарського засобу та вiдтворюваностi методiв контролю проводяться вiдповiдно до специфiкацiї та методiв контролю, наданих у матерiалах реєстрацiйного досьє.
5.1. Додатковi випробування щодо пiдтвердження якостi лiкарського засобу не проводяться у таких випадках:
пiсля проведення клiнiчних випробувань (дослiдження еквiвалентностi) за умови, що контроль якостi вже проводився на дослiджуваному лiкарському засобi i специфiкацiя, методи контролю якостi та технологiя виробництва лiкарського засобу не зазнали змiн;
при реєстрацiї додаткових доз лiкарського засобу до вже зареєстрованого дозування за умови, що склад дозувань - пропорцiйноподiбний, виробництво лiкарського засобу вiдбувається на одному й тому самому обладнаннi в умовах, передбачених лiцензiєю на виробництво цього лiкарського засобу, проведено дослiдження та надано звiт з валiдацiї методiв контролю та технологiчного процесу щодо запропонованого додаткового дозування;
при одночаснiй реєстрацiї декiлькох дозувань лiкарського засобу випробування з якостi проводять лише для одного з дозувань за умови, що склад дозувань - пропорцiйноподiбний, виробництво лiкарського засобу вiдбувається на одному й тому самому обладнаннi в умовах, передбачених лiцензiєю на виробництво цього лiкарського засобу;
лiкарський засiб має високу вартiсть (еквiвалентну 500 i бiльше євро), при цьому постачання в Україну вiдбувається в обмежених об'ємах. У такому випадку контроль за показниками якостi проводиться за процедурою "Lot Release" (шляхом експертизи матерiалiв контролю якостi серiї лiкарського засобу (протоколи контролю якостi серiї, аналiтичнi звiти), наданих виробником) (далi - "Lot Release");
якщо для проведення випробування за окремими показниками якостi (наприклад, "Стерильнiсть", "Мiкробiологiчна чистота" тощо) необхiдна велика кiлькiсть зразкiв, а вартiсть одного зразка - еквiвалентна 100 i бiльше євро. У такому випадку контроль за цими показниками якостi проводиться за процедурою "Lot Release";
якщо для проведення випробування за окремими показниками якостi у жоднiй з уповноважених лабораторiй вiдсутнє необхiдне обладнання для проведення контролю за такими показниками. У такому випадку контроль за цими показниками якостi проводиться за процедурою "Lot Release";
якщо лiкарський засiб є лiкарським засобом обмеженого застосування (препарат-сирота). У такому випадку контроль за показниками якостi проводиться за процедурою "Lot Release";
при реєстрацiї лiкарського засобу пiд iншою назвою. У такому випадку до матерiалiв реєстрацiйного досьє долучають результати лабораторного контролю якостi, проведеного при реєстрацiї цього лiкарського засобу з попередньою назвою;
при перереєстрацiї лiкарського засобу, якщо протягом дiї його реєстрацiйного посвiдчення специфiкацiя та методи контролю якостi лiкарського засобу не змiнювались i заявником не вносились та не вносяться змiни до матерiалiв реєстрацiйного досьє у частинi, що стосується специфiкацiї та методiв контролю якостi лiкарського засобу;
при реєстрацiї (перереєстрацiї) АФI або дiючих речовин;
для лiкарських засобiв, виготовлених з лiкарського засобу у формi in bulk шляхом фасування (пакування)/маркування, за умови, що для лiкарського засобу у формi in bulk було проведено передреєстрацiйний контроль якостi. У такому випадку до матерiалiв реєстрацiйного досьє долучаються результати лабораторного контролю якостi лiкарського засобу у формi in bulk;
при внесеннi змiн до iнструкцiї для медичного застосування лiкарського засобу (змiни терапевтичних показань, дозуючого пристрою тощо);
при реєстрацiї лiкарського засобу за процедурою "заява iнформованої згоди" за умови наявностi на ринку України власника реєстрацiйного посвiдчення зареєстрованого ранiше лiкарського засобу. У такому випадку до матерiалiв реєстрацiйного досьє долучаються результати лабораторного контролю якостi зареєстрованого ранiше лiкарського засобу.
5.2. Додатковi випробування з контролю активностi антибiотикiв не проводяться у таких випадках:
у матерiалах реєстрацiйного досьє наведено власнi дослiдження in vitro з контролю активностi антибiотикiв;
у матерiалах реєстрацiйного досьє представлено доказ терапевтичної еквiвалентностi лiкарського засобу шляхом проведення клiнiчних випробувань.
5.3. При реєстрацiї та перереєстрацiї медичного iмунобiологiчного препарату проводиться контроль однiєї серiї цього препарату на вiдповiднiсть вимогам МКЯ.
6. Звiти щодо проведених додаткових випробувань заявник надає до Центру для проведення спецiалiзованої експертизи.
XXI. ПОРЯДОК ВИДАЧI РЕЄСТРАЦIЙНОГО ПОСВIДЧЕННЯ (ВКЛАДКИ ДО РЕЄСТРАЦIЙНОГО ПОСВIДЧЕННЯ) НА ЛIКАРСЬКИЙ ЗАСIБ
1. У разi складання Центром умотивованих висновкiв щодо ефективностi, безпеки та якостi, Центр протягом 10 робочих днiв готує та надає заявнику для редакцiйного узгодження проекти таких документiв:
при державнiй реєстрацiї (перереєстрацiї) лiкарського засобу: реєстрацiйне посвiдчення, iнструкцiї для медичного застосування лiкарського засобу та короткої характеристики лiкарського засобу (за її наявностi), МКЯ, що мiстить маркування первинної, вторинної (за наявностi) упаковок лiкарського засобу;
при внесеннi змiн до матерiалiв реєстрацiйного досьє протягом дiї реєстрацiйного посвiдчення на лiкарськiй засiб: вкладки до реєстрацiйного посвiдчення, змiн та оновленої редакцiї iнструкцiї для медичного застосування лiкарського засобу та короткої характеристики лiкарського засобу, структуру якої наведено у додатку 14 до цього Порядку (за її наявностi), змiн та оновленої редакцiї МКЯ, що мiстить маркування первинної, вторинної (за наявностi) упаковок лiкарського засобу.
Iнформацiя про видачу заявнику для редакцiйного узгодження проектiв документiв вноситься до електронної бази даних.
Якщо заявник не узгоджує редакцiю проектiв документiв протягом 15 календарних днiв, Центр передає цi документи до МОЗ.
Час, необхiдний для узгодження заявником редакцiї документiв, не входить до строку проведення експертизи матерiалiв реєстрацiйного досьє.
2. Протягом 3 робочих днiв пiсля прийняття МОЗ рiшення про державну реєстрацiю (перереєстрацiю) лiкарського засобу або про внесення змiн до матерiалiв реєстрацiйного досьє протягом дiї реєстрацiйного посвiдчення Центр повiдомляє заявника про прийняте рiшення та передає узгодженi iз заявником реєстрацiйнi посвiдчення або вкладки до реєстрацiйних посвiдчень у МОЗ для їх засвiдчення пiдписом керiвника вiдповiдального структурного пiдроздiлу МОЗ.
3. Реєстрацiйне посвiдчення (вкладка до реєстрацiйного посвiдчення) протягом трьох робочих днiв з дати отримання пiдписується керiвником вiдповiдального структурного пiдроздiлу МОЗ та засвiдчується печаткою. Оформленi реєстрацiйнi посвiдчення (вкладка до реєстрацiйного посвiдчення) разом iз затвердженими МОЗ реєстрацiйними документами (iнструкцiєю для медичного застосування лiкарського засобу або змiнами до неї; короткою характеристикою лiкарського засобу (за наявностi - на бажання заявника); фармакопейною статтею або МКЯ чи змiнами до них, що мiстять маркування первинної, вторинної (за наявностi) упаковок на лiкарський засiб) передаються для видачi заявнику за принципом "єдиного вiкна".
4. Реєстрацiйне посвiдчення iз зазначенням строку, протягом якого дозволяється застосування лiкарського засобу в Українi (вкладка до реєстрацiйного посвiдчення), видається МОЗ у мiру звернення заявникiв за наявностi доручення вiд заявника на отримання зазначених документiв.
XXII. ПОРЯДОК РОЗРАХУНКIВ
1. Оплатi пiдлягають попередня експертиза та спецiалiзована експертиза матерiалiв реєстрацiйного досьє для державної реєстрацiї (перереєстрацiї), внесення змiн до матерiалiв реєстрацiйного досьє протягом дiї реєстрацiйного посвiдчення лiкарського засобу, додатковi вивчення (випробування).
2. Оплата вищезазначених робiт проводиться вiдповiдно до умов укладеного договору мiж заявником та Центром або мiж заявником та закладом охорони здоров'я/установою, що проводить такi вивчення (випробування).
XXIII. ЗАХИСТ КОНФIДЕНЦIЙНОЇ IНФОРМАЦIЇ
1. Пiд час проведення експертизи матерiалiв на лiкарськi засоби, що подаються на державну реєстрацiю (перереєстрацiю), а також експертизи матерiалiв про внесення змiн до реєстрацiйних матерiалiв протягом дiї реєстрацiйного посвiдчення Центр зобов'язаний забезпечити захист конфiденцiйної реєстрацiйної iнформацiї вiд розголошення i недобросовiсного комерцiйного використання.
2. Не допускається ознайомлення третiх осiб з конфiденцiйною реєстрацiйною iнформацiєю, зняття копiй з таких матерiалiв на паперових, електронних або iнших носiях без письмової згоди власника такої iнформацiї чи в iнших випадках, встановлених чинним законодавством.
3. Обiг документiв, що мiстять конфiденцiйну реєстрацiйну iнформацiю, здiйснюється в Центрi.
4. Не допускаються до роботи з документами, що мiстять конфiденцiйну реєстрацiйну iнформацiю, особи, якi можуть мати конфлiкт iнтересiв iз заявником.
| Начальник Управлiння лiкарських засобiв та медичної продукцiї МОЗ України | Л. В. Коношевич |
| Додаток 1 до Порядку проведення експертизи реєстрацiйних матерiалiв на лiкарськi засоби, що подаються на державну реєстрацiю (перереєстрацiю), а також експертизи матерiалiв про внесення змiн до реєстрацiйних матерiалiв протягом дiї реєстрацiйного посвiдчення |
ЗАЯВА
про державну реєстрацiю лiкарського засобу
| Дата надходження заяви "___" ____________ 20__ року |
N ______________________ |
|
Я гарантую достовiрнiсть та вiдповiдаю за iнформацiю, що мiститься у наданих матерiалах реєстрацiйного досьє, вiдповiдно до додатка ___.
Згоден, що у разi ненадання матерiалiв реєстрацiйного досьє протягом трьох мiсяцiв вiдповiдно до додатка ___ у розглядi цiєї заяви буде вiдмовлено.
Усi данi одержано заявником в установленому законодавством порядку i не порушують права третьої сторони, захищенi патентом, свiдоцтвом про знак для товарiв та послуг (пункти 4.16 - 4.18 цього додатка).
Також цим пiдтверджується, що всi передбаченi збори сплачено/буде сплачено вiдповiдно до вимог законодавства.
р Доручення для ведення переговорiв/пiдписання документiв вiд iменi заявника додається (пункт 4.4 цього додатка).
Вiд iменi заявника
_____________________________________
(пiдпис)
_____________________________________
(iм'я та прiзвище)
_____________________________________
(посада)
М. П
1. ЗАГАЛЬНI ПУНКТИ ЗАЯВИ ПРО ДЕРЖАВНУ РЕЄСТРАЦIЮ
1.1. Ця заява подається вiдповiдно до такого:
Примiтка. Роздiл повинен бути заповнений для будь-якої заяви, уключаючи заяви, на якi є посилання у цьому роздiлi.
У разi надання заяви на вiдповiдний тип вилучають перелiк iнших видiв заяв, на якi не поширюється дана заява.
р ПОВНА I НЕЗАЛЕЖНА ЗАЯВА/АВТОНОМНА ЗАЯВА
| р Медичний
iмунобiологiчний препарат (абзаци сорок п'ятий та сорок шостий роздiлу II Порядку) |
р Iнший лiкарський засiб |
(надається реєстрацiйне досьє з адмiнiстративними, фармацевтичними, доклiнiчними та клiнiчними даними) |
|
р Нова дiюча речовина (далi - ДР)
Примiтка. ДР не зареєстрована в Українi.
р Вiдома ДР
Примiтка. ДР була зареєстрована ранiше.
р ЗАЯВА НА ЛIКАРСЬКИЙ ЗАСIБ, ЯКИЙ МАЄ ДОБРЕ ВИВЧЕНЕ МЕДИЧНЕ ЗАСТОСУВАННЯ
(надається реєстрацiйне досьє з повними адмiнiстративними, фармацевтичними даними та докладною науковою бiблiографiєю, що свiдчить про ефективнiсть та безпеку дiючої речовини (згiдно з вимогами роздiлiв IX та X Порядку).
р ЗАЯВА IНФОРМОВАНОЇ ЗГОДИ
(надається реєстрацiйне досьє з адмiнiстративними, фармацевтичними, доклiнiчними та клiнiчними даними разом зi згодою власника реєстрацiйного посвiдчення зареєстрованого ранiше лiкарського засобу використовувати його фармацевтичнi, доклiнiчнi та клiнiчнi данi)
| р Медичний
iмунобiологiчний препарат (абзаци сорок п'ятий та сорок шостий роздiлу II Порядку) |
р Iнший лiкарський засiб |
Примiтка. Зареєстрований лiкарський засiб i заява iнформованої згоди можуть мати як одного, так i рiзного(их) власника(iв) реєстрацiйного посвiдчення.
Зареєстрований лiкарський засiб:
| назва лiкарського засобу, сила дiї, лiкарська форма | ||
| власник реєстрацiйного посвiдчення | ||
| номер(и) реєстрацiйного посвiдчення |
р Додайте iнформовану згоду власника реєстрацiйного посвiдчення на зареєстрований лiкарський засiб (пункт 4.2 цього додатка).
р ЗАЯВА НА ГЕНЕРИЧНИЙ ЛIКАРСЬКИЙ ЗАСIБ
| р Однокомпонентний | р Багатокомпонентний |
(надається реєстрацiйне досьє з адмiнiстративними, фармацевтичними даними, уключаючи еквiвалентнiсть до референтного лiкарського засобу, доклiнiчними (лiтературний огляд) та клiнiчними (власнi данi або лiтературний огляд*) даними (згiдно з вимогами роздiлiв IX та X Порядку) |
|
____________
* Залежно вiд лiкарської форми.
Референтний лiкарський засiб:
| назва лiкарського засобу, сила дiї, лiкарська форма | ||
| власник реєстрацiйного посвiдчення | ||
| номер(и) реєстрацiйного посвiдчення |
Лiкарський засiб, що використовувався у дослiдженнях еквiвалентностi (якщо такi проводилися):
| назва лiкарського засобу, сила дiї, лiкарська форма | ||
| власник реєстрацiйного посвiдчення | ||
| номер(и) реєстрацiйного посвiдчення |
Примiтка. Роздiл необхiдно заповнювати для кожного лiкарського засобу, що використовувався у дослiдженнях еквiвалентностi.
р ЗАЯВА НА ПОДIБНИЙ БIОЛОГIЧНИЙ ЛIКАРСЬКИЙ ЗАСIБ (БIОСИМIЛЯР)
| р Медичний
iмунобiологiчний препарат (абзаци сорок п'ятий та сорок шостий роздiлу II Порядку) |
р Iнший лiкарський засiб |
(надається реєстрацiйне досьє з повними адмiнiстративними, фармацевтичними, хiмiчними, бiологiчними даними та вiдповiднi данi, що пiдтверджують належний рiвень безпеки (токсикологiчнi чи iншi доклiнiчнi данi) та ефективностi (клiнiчнi данi (згiдно з вимогами роздiлiв IX та X Порядку). |
|
Референтний бiологiчний лiкарський засiб:
| назва лiкарського засобу, сила дiї, лiкарська форма | ||
| власник реєстрацiйного посвiдчення | ||
| номер(и) реєстрацiйного посвiдчення |
Вiдмiнностi порiвняно iз референтним бiологiчним лiкарським засобом (якщо такi є):
р вiдмiнностi у вихiдному(их) матерiалi(ах);
р вiдмiнностi у виробничому процесi;
р iнше терапевтичне застосування;
р вiдмiнностi у лiкарськiй формi;
р iнша(i) сила(и) дiї (кiлькiснi змiни ДР);
р iнший спосiб(оби) введення;
| р iншi вiдмiнностi | |
|
| |
р ГIБРИДНА ЗАЯВА
(у випадках, коли лiкарський засiб не пiдпадає пiд визначення генеричного лiкарського засобу, або коли бiоеквiвалентнiсть не може бути продемонстрована пiд час дослiджень бiодоступностi, або у випадку вiдмiнностей у дiючiй речовинi, дозуваннi, лiкарськiй формi, шляху введення тощо надаються матерiали реєстрацiйного досьє з повними адмiнiстративними, фармацевтичними даними та Модулi 4 та 5, у яких мiстяться звiти про обмеженi доклiнiчнi дослiдження та клiнiчнi випробування, що проводилися заявником, i бiблiографiчнi посилання).
Зареєстрований лiкарський засiб:
| назва лiкарського засобу, сила дiї, лiкарська форма | ||
| власник реєстрацiйного посвiдчення | ||
| номер(и) реєстрацiйного посвiдчення |
Вiдмiнностi порiвняно iз зареєстрованим лiкарським засобом:
р iнша лiкарська форма;
р iнша(i) сила(и) дiї (кiлькiснi змiни ДР);
р iнший спосiб(оби) введення;
р iнша фармакокiнетика (уключаючи iншу бiодоступнiсть);
р iнше терапевтичне застосування;
| р iншi вiдмiнностi | |
|
| |
р ЗАЯВА НА ФIКСОВАНУ КОМБIНАЦIЮ
(надається реєстрацiйне досьє з адмiнiстративними, фармацевтичними, доклiнiчними та клiнiчними даними тiльки щодо цiєї комбiнацiї (згiдно з вимогами роздiлiв IX та X Порядку).
р ЗАЯВА НА ЛIКАРСЬКИЙ ЗАСIБ З ПРОДУКЦIЇ IN BULK
| р Медичний
iмунобiологiчний препарат (абзаци сорок п'ятий та сорок шостий роздiлу II Порядку) |
р Iнший лiкарський засiб |
(надається реєстрацiйне досьє з адмiнiстративними, фармацевтичними (виробника продукцiї in bulk, доповненими документацiєю виробника щодо стадiй, якi ним виконуються), доклiнiчними (виробника продукцiї in bulk) та клiнiчними (виробника продукцiї in bulk) даними (згiдно з вимогами роздiлiв IX та X Порядку) |
|
р ЗАЯВА НА ЗМIНИ, ЩО ПОТРЕБУЮТЬ НОВОЇ РЕЄСТРАЦIЇ
(надаються вiдповiднi роздiли матерiалiв реєстрацiйного досьє, якi обґрунтовують указанi змiни i є достатнiми для проведення експертизи)
Позначити необхiдне:
• Змiни активних речовин:
р замiна активної речовини на iншу сiль, ефiр, похiдну тощо за умови, що характеристики, якi визначають спiввiдношення користь/ризик, суттєво не вiдрiзняються вiд зареєстрованих;
р використання iнших iзомерiв або змiна iзомерного складу;
р незначнi змiни молекулярної структури речовин бiологiчного походження за винятком змiни в активнiй речовинi вакцин для профiлактики грипу;
р змiна вектора, який використовується для виробництва антигена або вихiдного матерiалу, включаючи новий головний банк клiтин вiд iншого джерела, якщо характеристики, якi визначають спiввiдношення користь/ризик, суттєво не вiдрiзняються вiд зареєстрованих;
р новий лiганд або механiзм з'єднання для радiофармацевтичного лiкарського засобу, якщо характеристики, якi визначають спiввiдношення користь/ризик, суттєво не вiдрiзняються вiд зареєстрованих;
р застосування iнших екстрагентiв або спiввiдношення рослинна лiкарська сировина/рослинний препарат для рослинного лiкарського засобу, якщо характеристики, якi визначають спiввiдношення користь/ризик, суттєво не вiдрiзняються вiд зареєстрованих.
• Змiни сили дiї, лiкарської форми та способу застосування:
р змiна бiодоступностi;
р змiна фармакокiнетики;
р змiна або введення додаткового дозування лiкарського засобу (додаткова доза);
р змiна або додавання нової лiкарської форми;
р змiна або додавання нового шляху введення (для парентеральних форм у зв'язку з вiдмiнностями в ефективностi та безпецi лiкарського засобу при внутрiшньоартерiальному, внутрiшньовенному, внутрiшньом'язовому та iнших шляхах введення).
Лiкарський засiб, зареєстрований в Українi, до якого вносяться вiдповiднi змiни
| назва лiкарського засобу, сила дiї, лiкарська форма | ||
| власник реєстрацiйного посвiдчення | ||
| номер(и) реєстрацiйного посвiдчення |
р ЗАЯВА НА ПРЕПАРАТ ОБМЕЖЕНОГО ЗАСТОСУВАННЯ (ПРЕПАРАТ-СИРОТА)
| р Медичний
iмунобiологiчний препарат (абзаци сорок п'ятий та сорок шостий роздiлу II Порядку) |
р Iнший лiкарський засiб |
1. Чи надано лiкарському засобу статус препарату обмеженого застосування (препарату-сироти)
р Нi
р У процесi розгляду
р Так
| дата | ||
| реєстрацiйний номер препарату обмеженого застосування (препарату-сироти) |
р Вiдмовлено у присвоєннi статусу препарату обмеженого застосування (препарату-сироти)
| дата | ||
| номер рiшення |
р Заяву на присвоєння статусу вiдкликано
| дата |
р Додайте копiю рiшення щодо присвоєння статусу препарату обмеженого застосування (препарату-сироти) (якщо є) (пункт 4.15 цього додатка).
2. ОСОБЛИВI ПУНКТИ ЗАЯВИ ПРО ДЕРЖАВНУ РЕЄСТРАЦIЮ
2.1. Назва та код АТХ
| 2.1.1. Назва лiкарського засобу |
| 2.1.2. Назва ДР або склад (указати) Примiтка. Тiльки одна назва повинна бути наведена в такому порядку: Мiжнародна непатентована назва (далi - МНН)*, Державна фармакопея України (далi - ДФУ), Європейська фармакопея, загальноприйнята назва, наукова (хiмiчна) назва. ____________ |
2.1.3. Фармакотерапевтична група
(використовуйте дiючий код АТХ)
Якщо код АТХ не присвоєно, зазначте, чи була подана заява на присвоєння коду АТХ р Малюнок 1 |
2.2. Сила дiї (дозування), лiкарська форма та упаковка, шлях введення, об'єм контейнера та розмiр упаковки
| 2.2.1. Сила дiї (дозування) i лiкарська
форма (використовуйте перелiк стандартних термiнiв ДФУ або Європейської фармакопеї)
|
| 2.2.2. Шлях(и) введення (використовуйте
перелiк стандартних термiнiв ДФУ або Європейської
фармакопеї) |
| 2.2.3. Упаковка: контейнер,
укупорювальна система та пристрої для введення, уключаючи
опис матерiалу, з якого вони виготовленi
(використовуйте перелiк стандартних термiнiв ДФУ
або Європейської фармакопеї) Для кожного виду упаковки вкажiть: 2.2.3.1. Розмiр(и) упаковки. 2.2.3.2. Пропонований термiн придатностi. 2.2.3.3. Пропонований термiн придатностi (пiсля першого розкриття упаковки/контейнера). 2.2.3.4. Пропонований термiн придатностi (пiсля вiдновлення/розчинення або розведення). 2.2.3.5. Пропонованi умови зберiгання. 2.2.3.6. Пропонованi умови зберiгання пiсля першого розкриття упаковки. р Додайте пропозицiї до маркування на упаковцi (пункт 4.14 цього додатка) |
2.3. Правовий статус
| 2.3.1. Пропонована категорiя вiдпуску: р за рецептом р без рецепта р тiльки в умовах стацiонару |
| 2.3.2. Для лiкарських засобiв, пропонованих для рецептурного вiдпуску, заявник надає свої пропозицiї щодо категорiї вiдпуску лiкарського засобу, однак право визначати категорiю вiдпуску залишається за МОЗ України з урахуванням вимог наказу Мiнiстерства охорони здоров'я України вiд 17 травня 2001 року N 185 "Про затвердження критерiїв визначення категорiй вiдпуску лiкарських засобiв", зареєстрованого у Мiнiстерствi юстицiї України 31 травня 2001 року за N 464/5655. |
| 2.3.3. Пропозицiї щодо рекламування
лiкарських засобiв, що вiдпускаються без рецепта: р рекламування тiльки серед медичних працiвникiв р рекламування для населення та медичних працiвникiв |
2.4. Заявник (власник реєстрацiйного посвiдчення)/контактнi особи
2.4.1. Заявник (власник реєстрацiйного посвiдчення):
| найменування юридичної особи або прiзвище, iм'я, по батьковi фiзичної особи - пiдприємця | ||
| мiсцезнаходження юридичної особи або мiсце проживання фiзичної особи - пiдприємця | ||
| країна | ||
| телефон/факс | ||
2.4.2. Представник заявника
(уповноважена особа, що виступає вiд iменi
заявника):
р У разi заповнення цього пункту додайте доручення (пункт 4.4 цього додатка). |
||||||||||||||||||
2.4.3. Представник заявника
(уповноважена особа, що виступає вiд iменi
заявника) пiсля реєстрацiї лiкарського засобу,
якщо вони вiдмiннi вiд визначених у пунктi 2.4.2:
р У разi заповнення цього пункту додайте доручення (пункт 4.4 цього додатка). |
2.4.4. Уповноважена квалiфiкована особа
заявника, вiдповiдальна за фармаконагляд:
р Додайте бiографiчну довiдку уповноваженої квалiфiкованої особи, вiдповiдальної за фармаконагляд (пункт 4.5 цього додатка), та гарантiйний лист заявника (пункт 4.21 цього додатка). Укажiть мiсце проживання уповноваженої квалiфiкованої особи заявника, вiдповiдальної за фармаконагляд 2.4.5. Уповноважена квалiфiкована особа заявника в Українi для здiйснення фармаконагляду, якщо вона вiдмiнна вiд зазначеної у пунктi 2.4.4:
р Якщо вiдрiзняється вiд пункту 2.4.4, додайте бiографiчну довiдку уповноваженої квалiфiкованої особи в Українi для здiйснення фармаконагляду (пункт 4.5 цього додатка). Укажiть мiсце проживання уповноваженої квалiфiкованої особи в Українi для здiйснення фармаконагляду |
2.5. Виробники
Усi данi щодо виробничих дiльниць, на яких здiйснюються виробничi процедури, та дiльниць, на яких проводиться контроль якостi, що зазначаються у матерiалах реєстрацiйного досьє, мають збiгатися щодо їх найменувань, мiсцезнаходжень та видiв дiяльностi.
2.5.1. Виробник(и), що вiдповiдає(ють) за
випуск серiї (як указано в iнструкцiї про
застосування лiкарського засобу (iнструкцiї для
медичного застосування) та, за необхiдностi, у
маркуваннi)
р Додайте копiю лiцензiї на виробництво (пункт 4.6 цього додатка). р Додайте копiю сертифiката вiдповiдностi вимогам належної виробничої практики, передбаченого у пунктi 4.8 цього додатка. 2.5.2. Лабораторiя країни виробника з контролю якостi препаратiв кровi та вакцин, вiдповiдальна за контроль якостi/випуск серiї
|
2.5.3. Контактна особа, вiдповiдальна за
роботу з рекламацiями щодо дефектної продукцiї
|
| 2.5.4. Виробник(и) лiкарського засобу i
дiльниця(i) виробництва: Включаючи дiльницi виробництва будь-якого розрiджувача/розчинника в окремiй упаковцi/ємностi, що є частиною лiкарського засобу, дiльницi, на яких здiйснюється контроль якостi/контроль у процесi виробництва, а також (за потреби) iмпортерiв
р Додайте технологiчну схему виробництва (пункт 4.7 цього додатка). р Додайте копiю сертифiката вiдповiдностi вимогам належної виробничої практики, передбаченого у пунктi 4.8 цього додатка. р Додайте копiю лiцензiї на виробництво (пункт 4.6 цього додатка). Зазначте прiзвище та iм'я уповноваженої особи виробника (якщо не вказано у лiцензiї на виробництво) Чи проводилася перевiрка дiльницi на дотримання вимог належної виробничої практики (далi - GMP) уповноваженим органом України або органами країн вiдповiдно до мiжнародних договорiв про взаємне визнання результатiв iнспектування на вiдповiднiсть виробництва вимогам GMP?
Якщо так, укажiть:
висновок:
р Додайте для кожної дiльницi виробництва висновок про вiдповiднiсть вимогам GMP вiдповiдно до пункту 4.8 цього додатка. |
| 2.5.5. Виробник(и) ДР та виробничi дiльницi Повиннi бути зазначенi всi дiльницi виробництва, задiянi у виробничому процесi кожного джерела дiючої речовини, включаючи дiльницi, на яких проводиться контроль якостi/контроль у процесi виробництва. Для високотехнологiчних (бiотехнологiчних) лiкарських засобiв мають бути включенi усi дiльницi зберiгання головного та робочого банкiв клiтин та дiльницi приготування робочого банку клiтин.
Короткий опис етапiв виробництва, виконуваних на виробничiй дiльницi р Додайте технологiчну схему виробництва (пункт 4.7 цього додатка). р Для кожної ДР додайте заяву уповноваженої особи виробника згiдно з пунктом 4.22 цього додатка. Чи проводилася перевiрка дiльницi на дотримання вимог GMP уповноваженим органом України або органами країн вiдповiдно до мiжнародних договорiв про взаємне визнання результатiв iнспектування на вiдповiднiсть виробництва вимогам GMP?
р Додайте для кожної дiльницi виробництва документ, передбачений пунктом 4.6 цього додатка, та, за наявностi, р Додайте копiю сертифiката вiдповiдностi вимогам належної виробничої практики вiдповiдно до пункту 4.8 цього додатка. · Чи видано сертифiкат вiдповiдностi Європейськiй фармакопеї для ДР?
Якщо так:
р Додайте копiю сертифiката вiдповiдностi (пункт 4.9 цього додатка). · Чи є матерiали реєстрацiйного досьє (мастер-файл) на ДР, посилання на якi буде використовуватися у досьє, оригiнальними?
Якщо так:
р Додайте лист(и) виробника(iв) ДР, що дає(ють) право використання мастер-файла на ДР (пункт 4.9 цього додатка). р Додайте копiю письмового зобов'язання вiд виробника(iв) ДР iнформувати заявника про змiни у виробничому процесi або специфiкацiях (пункт 4.10 цього додатка). · Чи видано сертифiкат на загальний файл на вакцинний антиген (далi - ВАЗФ), що використовується у цiй заявi, або на нього подано заявку?
Якщо так:
р Додайте копiю сертифiката на ВАЗФ (пункт 4.19 цього додатка) |
| 2.5.6. Заклади охорони здоров'я/установи,
що залучалися до клiнiчного(их) випробування(нь)
для дослiдження бiодоступностi та/або
бiоеквiвалентностi, або такi, що залучалися для
валiдацiї процесiв виробництва продуктiв кровi Для кожного закладу охорони здоров'я/установи вкажiть, де проводилися аналiтичнi тестування та де зiбрано клiнiчнi данi, а також:
|
2.6. Якiсний та кiлькiсний склад лiкарського засобу
| 2.6.1. Якiсний та кiлькiсний склад
лiкарського засобу (ДР та допомiжнi речовини): Необхiдно вказати, на яку кiлькiсть розраховано склад (наприклад, 1 капсула). Перерахуйте дiючi речовини окремо вiд допомiжних речовин:
Примiтка. * Тiльки одну назву для
кожної ДР необхiдно вказати в такiй послiдовностi:
МНН**, ДФУ, Європейська фармакопея,
загальноприйнята назва, наукова (хiмiчна) назва. Iнформацiю про надлишкову кiлькiсть не слiд указувати в колонках щодо складу, а необхiдно викласти нижче:
|
| 2.6.2. Перелiк матерiалiв тваринного
та/або людського походження, що входять до складу
або використовуються у процесi виробництва
лiкарського засобу ВIДСУТНI р
р Якщо є сертифiкат вiдповiдностi ЄФ5 щодо ГЕ4 або документ, що видано уповноваженими органами ветеринарного нагляду країни походження сировини щодо реєстрацiї в країнi (за результатами клiнiчного та лабораторного контролю) випадкiв ГЕ4, вiн повинен бути доданий у пунктi 4.11 цього додатка. ____________ |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||
2.6.3. Чи видано сертифiкат на матерiали
реєстрацiйного досьє (мастер-файл) на плазму (далi
- ПМФ), що використовується у цiй заявi на
реєстрацiю, або на нього подано заявку?
Якщо так:
Функцiя
____________ р Додайте копiю сертифiката (пункт 4.20 цього додатка). |
2.6.4. Чи мiстить або складається
лiкарський засiб з генетично модифiкованих
органiзмiв (ГМО)?
Якщо так, то чи вiдповiдає лiкарський засiб встановленим вимогам? Зробiть необхiдне посилання
р Додайте копiю письмового дозволу, виданого уповноваженим органом країни виробника, на навмисне вивiльнення ГМО у навколишнє середовище при використаннi з дослiдницькою метою (пункт 4.12 цього додатка). |
3. IНШI ВIДОМОСТI
3.1. Чи захищено лiкарський засiб
патентами на винахiд, корисну модель або
промисловий зразок, дiя яких розповсюджується на
Україну?
Якщо так, то наведiть таку iнформацiю:
р Додайте копiї патентiв згiдно з пунктом 4.16 цього додатка. р Для державної реєстрацiї лiкарських засобiв, що базуються або мають вiдношення до об'єктiв iнтелектуальної власностi, на якi вiдповiдно до законiв України видано патент, заявник подає засвiдчену вiдповiдно копiю патенту або лiцензiї, якою дозволяється виробництво та продаж зареєстрованого лiкарського засобу, а також документ, що пiдтверджує чиннiсть патенту в Українi. Заявники подають лист, в якому вказується, що права третьої сторони, захищенi патентом або переданi за лiцензiєю, не порушуються у зв'язку з реєстрацiєю лiкарського засобу. |
3.2. Чи захищена торговельна марка в
Українi?
Якщо так, то наведiть таку iнформацiю:
р Додайте копiї документiв, що передбаченi в пунктi 4.17 цього додатка. |
3.3. Чи зареєстровано лiкарський засiб у
країнi виробника та iнших країнах?
Якщо так: р Додайте копiю реєстрацiйного посвiдчення (пункт 4.3 цього додатка). р Зазначте перелiк країн, де лiкарський засiб зареєстрований/перереєстрований. |
3.4. Чи була проведена попередня
наукова консультацiя щодо цього лiкарського
засобу в Українi?
Якщо так:
Чи була проведена попередня наукова консультацiя щодо цього лiкарського засобу в iншiй країнi?
Якщо так:
р Додайте копiю консультацiйного листа (пункт 4.13 цього додатка). |
4. ДОКУМЕНТИ, ЩО ДОДАЮТЬСЯ (позначте необхiдне)
р 4.1. Документ, що пiдтверджує сплату реєстрацiйного збору.
р 4.2. Iнформована згода власника реєстрацiйного посвiдчення на зареєстрований лiкарський засiб (у довiльнiй формi).
р 4.3. Копiя реєстрацiйного посвiдчення або iншого дозвiльного документа (Marketing Authorization, Certificate of a Pharmaceutical Product, Free Sale Certificate тощо), виданого уповноваженим органом країни власника реєстрацiйного посвiдчення (заявника) та/або виробника, або iншої країни, регуляторний орган якої керується високими стандартами до якостi, що вiдповiдають стандартам, рекомендованим ВООЗ, на ринку якої розмiщено лiкарський засiб, iз зазначенням назви документа та назви компетентного органу, що його видав, дати видачi i сторiнки, пiдписаної уповноваженою особою компетентного органу (для продукцiї in bulk - пiдтвердження реєстрацiї лiкарського засобу, виготовленого iз заявленої продукцiї in bulk, у споживчiй упаковцi в країнi виробника або iншiй країнi, регуляторний орган якої керується високими стандартами до якостi, що вiдповiдають стандартам, рекомендованим ВООЗ, на ринку якої розмiшено зазначений лiкарський засiб). Копiя має бути засвiдчена печаткою заявника/представника заявника в Українi. Зазначити перелiк країн, де даний лiкарський засiб зареєстровано/перереєстровано.
р 4.4. Доручення для ведення переговорiв/пiдписання документiв вiд iменi заявника (власника реєстрацiйного посвiдчення).
р 4.5. Бiографiчна довiдка уповноваженої квалiфiкованої особи, вiдповiдальної за здiйснення фармаконагляду (у довiльнiй формi).
р 4.6. Копiя лiцензiї на виробництво (якщо згiдно iз законодавством країни виробника лiцензiя на виробництво iснує лише в електронному виглядi (наприклад, США), має бути надана роздрукiвка iз посиланням на вiдповiдний офiцiйний сайт, засвiдчена пiдписом/печаткою заявника) або iншого дозвiльного документа на виробництво заявленої лiкарської форми у країнi виробника. Копiя повинна бути засвiдчена печаткою заявника (для фiзичної особи за наявностi)/представника заявника в Українi.
р 4.7. Проект технологiчного регламенту або вiдомостi про технологiю виробництва (технологiчна схема виробництв iз зазначенням усiх дiльниць виробництва, задiяних у процесi виробництва лiкарського засобу та дiючої речовини (уключаючи дiльницi для вiдбору зразкiв i контролю серiй лiкарського засобу). Копiя повинна бути засвiдчена печаткою заявника (для фiзичної особи за наявностi)/представника заявника в Українi.
р 4.8. Засвiдчена копiя документа, що пiдтверджує вiдповiднiсть виробництва лiкарського засобу вимогам GMP, виданого Держлiкслужбою вiдповiдно до вимог наказу Мiнiстерства охорони здоров'я України вiд 27 грудня 2012 року N 1130 "Про затвердження Порядку проведення пiдтвердження вiдповiдностi умов виробництва лiкарських засобiв вимогам належної виробничої практики", зареєстрованого у Мiнiстерствi юстицiї України 21 сiчня 2013 року за N 133/22665, або гарантiйний лист заявника щодо надання такого документа протягом строку проведення спецiалiзованої експертизи. За необхiдностi - висновки щодо iнших iнспекцiй, якi проводилися. Копiї документiв мають бути засвiдченi печаткою заявника (для фiзичної особи за наявностi)/представника заявника в Українi.
р 4.9. Письмова згода на використання матерiалiв реєстрацiйного досьє (мастер-файл) на дiючу речовину (у довiльнiй формi) вiд її виробника або копiя сертифiката вiдповiдностi Європейськiй фармакопеї.
р 4.10. Копiя письмового зобов'язання виробника дiючої речовини (у довiльнiй формi) iнформувати заявника про змiни у виробничому процесi або специфiкацiях.
р 4.11. Сертифiкат вiдповiдностi Європейськiй фармакопеї щодо губчатої енцефалопатiї або документ, виданий уповноваженими органами ветеринарного нагляду країни походження сировини, щодо реєстрацiї в країнi (за результатами клiнiчного та лабораторного контролю) випадкiв губчатої енцефалопатiї.
р 4.12. Копiя письмового дозволу, виданого уповноваженим органом країни виробника, на навмисне вивiльнення ГМО у навколишнє середовище при використаннi з дослiдницькою метою.
р 4.13. Копiя консультацiйного листа до проведеної попередньої наукової консультацiї щодо лiкарського засобу.
р 4.14. Пропозицiї до маркування упаковки лiкарського засобу.
р 4.15. Копiя рiшення про присвоєння статусу препарату обмеженого застосування (препарату-сироти).
р 4.16. Копiї патентiв на винахiд, корисну модель або промисловий зразок, дiя яких розповсюджується на Україну.
р 4.17. Копiї документiв щодо захисту торговельної марки в Українi.
р 4.18. Лист, форма якого наведена у додатку 15 до цього Порядку.
р 4.19. Копiя сертифiката на ВАЗФ.
р 4.20. Копiя сертифiката на ПМФ.
р 4.21. Гарантiйний лист заявника (у довiльнiй формi) щодо забезпечення функцiонування належної системи нагляду за безпекою лiкарських засобiв при їх медичному застосуваннi, у тому числi в Українi.
р 4.22. Для кожної дiючої речовини дiльниць виробництва, задiяних у виробничому процесi, включаючи дiльницi, на яких проводиться контроль якостi/контроль у процесi виробництва та випуск серiй, заява уповноваженої особи щодо вiдповiдностi виробництва вимогам належної виробничої практики та Керiвництвам з належної виробничої практики щодо вихiдних матерiалiв.
| Додаток 2 до Порядку проведення експертизи реєстрацiйних матерiалiв на лiкарськi засоби, що подаються на державну реєстрацiю (перереєстрацiю), а також експертизи матерiалiв про внесення змiн до реєстрацiйних матерiалiв протягом дiї реєстрацiйного посвiдчення |
Заява
про державну реєстрацiю (перереєстрацiю)
лiкарського засобу
| Дата надходження заяви "___" ____________ 20__ року |
N ________________ |
| 1. Назва лiкарського засобу |
| |
| |
| |
|
| |
|
| |
|
| 3. Упаковка: | |
| первинна | |
| вторинна | |
| 4. Заявник (власник реєстрацiйного
посвiдчення) (для вiтчизняних заявникiв -
українською, для зарубiжних - українською та
англiйською мовами). Найменування юридичної особи або прiзвище, iм'я, по батьковi фiзичної особи - пiдприємця |
| |
| |
| |
| |
| |
|
| |
|
| |
|
| Телефон/факс | |
| |
| Керiвник | |
| |
|
| 4.1. Представник заявника (уповноважена особа, що виступає вiд iменi заявника) (українською мовою): |
| |
|
| |
|
| |
|
| |
|
| |
|
| |
|
| |
|
| |
|
| |
|
| |
|
| |
|
| Телефон/факс | |
| |
| 4.2. Уповноважена квалiфiкована особа заявника для здiйснення фармаконагляду в Українi (українською мовою): |
| |
|
| |
|
| |
|
| Найменування юридичної особи або прiзвище, iм'я, по батьковi фiзичної особи - пiдприємця |
| (заявника) | |
| |
|
| Мiсцезнаходження юридичної особи або мiсце проживання фiзичної особи - пiдприємця |
| (заявника) | |
| |
|
| |
|
| |
|
| |
|
| Цiлодобовий телефон/факс | |
| |
| 5. Виробник(и) (для вiтчизняних виробникiв -
українською, для зарубiжних - українською та
англiйською мовами) 5.1. Виробник(и) лiкарського засобу Найменування юридичної особи або прiзвище, iм'я, по батьковi фiзичної особи - пiдприємця |
| |
| |
| |
|
| |
|
| |
|
| Телефон/факс | |
| |
| Керiвник | |
| |
|
| Виробництво лiкарського засобу (потрiбне позначити): | |
| р | повнiстю даним виробником |
| р | частково даним виробником |
| р | повнiстю iншим виробником |
| 5.2. Виробник(и) дiючої(их) речовини(н) | |
| Дiюча речовина | |
| |
|
| Найменування юридичної особи або прiзвище, iм'я, по батьковi фiзичної особи - пiдприємця |
| (виробника) | |
| |
|
| |
|
| |
|
| |
|
| |
|
| Телефон/факс | |
| |
| Керiвник | |
| |
|
| 6. Якiсний i кiлькiсний склад лiкарського
засобу (дiючi та допомiжнi речовини) Укажiть дiючу(i) речовину(и) окремо вiд допомiжних |
|
| Назва речовини* | Кiлькiсть на одиницю лiкарської форми** | Посилання/монографiя |
____________
* Повинна вказуватися тiльки
одна назва в такому порядку: мiжнародна
непатентована назва, Державна фармакопея
України, Європейська фармакопея,
загальноприйнята назва, наукова (хiмiчна) назва.
Дiюча речовина повинна бути заявлена за
рекомендованою мiжнародною непатентованою
назвою iз зазначенням форми солi або гiдрату, якщо
необхiдно.
** В одиницях ваги чи
бiологiчних одиницях на 1 одиницю лiкарської
форми: драже, таблетки, супозиторiї, ампули,
флакони; у вiдсотках чи мг/мл, мг/г: мазi, креми,
розчини, неподiльнi порошки, збори.
| 7. Фармакотерапевтична група |
| |
| |
| |
| 9. Захищенiсть патентами на винахiд, корисну модель або промисловий зразок, дiя яких розповсюджується на Україну (потрiбне зазначити): |
|
|
|||||
| Якщо так, то навести iнформацiю: | ||||||
| Номер патенту | Дата видачi | Дiє до | Власник патенту |
| 10. Захищенiсть торговельної марки в Українi
(потрiбне зазначити): Якщо так, то навести iнформацiю: |
|
|
| Номер патенту | Дата видачi | Дiє до | Власник патенту |
| |
| |
|
| |
|
| 13. Пропонована категорiя вiдпуску: р за рецептом р без рецепта р тiльки в умовах стацiонару Для лiкарських засобiв, пропонованих для рецептурного вiдпуску, заявник надає свої пропозицiї щодо категорiї вiдпуску лiкарського засобу, однак право визначати категорiю вiдпуску залишається за МОЗ України з урахуванням вимог наказу Мiнiстерства охорони здоров'я України вiд 17 травня 2001 року N 185 "Про затвердження критерiїв визначення категорiй вiдпуску лiкарських засобiв", зареєстрованого у Мiнiстерствi юстицiї України 31 травня 2001 року за N 464/5655. Заявник є вiдповiдальним за якiсть, безпеку та ефективнiсть лiкарського засобу в порядку, визначеному чинним законодавством, гарантує достовiрнiсть iнформацiї, що мiститься в матерiалах реєстрацiйного досьє, а також пiдтверджує, що в матерiалах реєстрацiйного досьє наявнi данi щодо якостi, безпеки та ефективностi лiкарського засобу. Усi данi одержано заявником в установленому порядку i права третьої сторони, захищенi патентом або переданi за лiцензiєю, не порушуються у зв'язку з реєстрацiєю лiкарського засобу. |
| ДОКУМЕНТИ, ЩО ДОДАЮТЬСЯ (позначте необхiдне): |
| р 1. Доручення для ведення
переговорiв/пiдписання документiв вiд iменi
заявника (власника реєстрацiйного посвiдчення). р 2. Копiя лiцензiї на виробництво (якщо згiдно iз законодавством країни виробника лiцензiя на виробництво iснує лише в електронному виглядi (наприклад, США), має бути надана роздрукiвка iз посиланням на вiдповiдний офiцiйний сайт, засвiдчена пiдписом/печаткою заявника (для фiзичної особи - за наявностi), або iншого дозвiльного документа на виробництво заявленої лiкарської форми у країнi виробника. Копiя повинна бути засвiдчена печаткою заявника (для фiзичної особи - за наявностi)/представника заявника в Українi. р 3. Копiї патентiв на винахiд, корисну модель або промисловий зразок, дiя яких розповсюджується на Україну. р 4. Копiї документiв щодо захисту торговельної марки в Українi. р Копiя реєстрацiйного посвiдчення або iншого дозвiльного документа (Marketing Authorization, Certificate of a Pharmaceutical Product, Free Sale Certificate тощо), виданого уповноваженим органом країни власника реєстрацiйного посвiдчення (заявника) та/або виробника, або iншої країни, регуляторний орган якої керується високими стандартами до якостi, що вiдповiдають стандартам, рекомендованим ВООЗ, на ринку якої розмiшено лiкарський засiб, iз зазначенням назви документа та назви компетентного органу, що його видав, дати видачi i сторiнки, пiдписаної уповноваженою особою компетентного органу. Копiя має бути засвiдчена печаткою заявника (для фiзичної особи - за наявностi)/представника заявника в Українi. Зазначити перелiк країн, де даний лiкарський засiб зареєстровано/перереєстровано. р 6. Копiї реєстрацiйного(их) посвiдчення(нь) та вкладки до них (у разi перереєстрацiї). р 7. Засвiдчена копiя документа, що пiдтверджує вiдповiднiсть виробництва лiкарського засобу вимогам GMP, виданого Держлiкслужбою вiдповiдно до вимог наказу Мiнiстерства охорони здоров'я України вiд 27 грудня 2012 року N 1130 "Про затвердження Порядку проведення пiдтвердження вiдповiдностi умов виробництва лiкарських засобiв вимогам належної виробничої практики", зареєстрованого у Мiнiстерствi юстицiї України 21 сiчня 2013 року за N 133/22665, або гарантiйний лист заявника щодо надання такого документа протягом строку проведення спецiалiзованої експертизи. За необхiдностi - висновки щодо iнших iнспекцiй, якi проводилися. Копiї документiв мають бути засвiдченi печаткою заявника (для фiзичної особи - за наявностi)/представника заявника в Українi. |
| Дата заповнення: "___" ____________ 20__ року |
Пiдпис керiвника заявника або
уповноваженої особи, що виступає вiд iменi
заявника ________________________ М. П. |
| Додаток 3 до Порядку проведення експертизи реєстрацiйних матерiалiв на лiкарськi засоби, що подаються на державну реєстрацiю (перереєстрацiю), а також експертизи матерiалiв про внесення змiн до реєстрацiйних матерiалiв протягом дiї реєстрацiйного посвiдчення |
ЗАЯВА
про державну реєстрацiю (перереєстрацiю) АФI або
дiючої речовини
| Дата надходження заяви "___" __________ 20__ року |
N ______________________ |
Назва повинна бути наведена в такому порядку: мiжнародна непатентована назва*, Державна фармакопея України (далi - ДФУ), Європейська фармакопея, загальноприйнята назва, наукова (хiмiчна) назва. ____________ |
Я гарантую достовiрнiсть та вiдповiдаю за
iнформацiю, що мiститься у наданих матерiалах
реєстрацiйного досьє.
Згоден, що у разi ненадання матерiалiв
реєстрацiйного досьє протягом трьох мiсяцiв у
розглядi цiєї заяви буде вiдмовлено.
Також цим пiдтверджується, що всi передбаченi
збори сплачено/буде сплачено вiдповiдно до вимог
законодавства.
р Додайте доручення для ведення
переговорiв/пiдписання документiв вiд iменi
заявника (пункт 3.2 цього додатка).
Вiд iменi заявника
_____________________________________
(пiдпис)
_____________________________________
(iм'я та прiзвище)
_____________________________________
(посада)
М. П.
1. ОСОБЛИВI ПУНКТИ ЗАЯВИ
| р Медичний
iмунобiологiчний препарат (абзаци сорок п'ятий та сорок шостий роздiлу III Порядку) |
р Iнший лiкарський засiб |
| 1.1. Технологiчна форма (форма випуску) (рiдина, порошок, гранули, пелети тощо) |
| 1.2. Упаковка, уключаючи опис
матерiалу, з якого вони виготовленi
(використовуйте перелiк стандартних термiнiв ДФУ
або Європейської фармакопеї) Для кожного виду упаковки укажiть: 1.2.1. Розмiр(и) упаковки. 1.2.2. Пропонований термiн придатностi. 1.2.3. Пропонований перiод проведення переконтролю. 1.2.4. Пропонованi умови зберiгання. |
1.3. Заявник (власник реєстрацiйного посвiдчення)/контактна особа
1.3.1. Заявник (власник реєстрацiйного
посвiдчення) (для вiтчизняних заявникiв -
українською, для зарубiжних - українською та
англiйською мовами):
|
1.3.2. Представник заявника
(уповноважена особа, що виступає вiд iменi
заявника):
р У разi заповнення цього пункту додайте доручення (пункт 3.2 цього додатка) |
1.4. Виробники
1.4.1. Виробник, що вiдповiдає за випуск
серiї АФI або дiючої речовини (для вiтчизняних
заявникiв - українською, для зарубiжних -
українською та англiйською мовами):
|
| 1.4.2. Виробник(и) та виробничi дiльницi (для
вiтчизняних заявникiв - українською, для
зарубiжних - українською та англiйською мовами). Повиннi бути зазначенi всi дiльницi виробництва, задiянi у виробничому процесi кожного джерела АФI або дiючої речовини, включаючи дiльницi, на яких проводиться контроль якостi/контроль у процесi виробництва. Для високотехнологiчних (бiотехнологiчних) лiкарських засобiв мають бути включенi усi дiльницi зберiгання головного та робочого банкiв клiтин i дiльницi приготування робочого банку клiтин.
Короткий опис етапiв виробництва, виконуваних на виробничiй дiльницi р Додайте технологiчну схему виробництва (пункт 3.4 цього додатка). · Чи видано сертифiкат вiдповiдностi Європейськiй фармакопеї для дiючої речовини (далi - ДР)?
Якщо так:
р Додайте копiю сертифiката вiдповiдностi (пункт 3.5 цього додатка). · Чи є матерiали реєстрацiйного досьє (мастер-файл) на ДР оригiнальними?
Якщо так:
р Для АФI або дiючих речовин тваринного походження, якщо є сертифiкат вiдповiдностi Європейськiй фармакопеї щодо губчатої енцефалопатiї (далi - ГЕ) або документ, виданий уповноваженими органами ветеринарного нагляду країни походження сировини щодо реєстрацiї в країнi (за результатами клiнiчного та лабораторного контролю) випадкiв ГЕ, вiн повинен бути доданий у пунктi 3.6 цього додатка. |
1.5. Чи мiстить або складається АФI або
дiюча речовина з генетично модифiкованих
органiзмiв (ГМО)?
Якщо так, то чи вiдповiдає АФI або дiюча речовина встановленим вимогам? Зробiть необхiдне посилання
р Додайте копiю письмового дозволу, виданого уповноваженим органом країни виробника, на навмисне вивiльнення ГМО у навколишнє середовище при використаннi з дослiдницькою метою (пункт 3.7 цього додатка). |
2. IНШI ВIДОМОСТI
2.1. Чи захищений АФI або дiюча речовина
патентами на винахiд, корисну модель або
промисловий зразок, дiя яких розповсюджується на
Україну?
Якщо так, то наведiть таку iнформацiю:
р Додайте копiї патентiв згiдно з пунктом 3.8 цього додатка. р Для державної реєстрацiї АФI або дiючих речовин, що базуються або мають вiдношення до об'єктiв iнтелектуальної власностi, на якi вiдповiдно до законiв України видано патент, заявник подає засвiдчену вiдповiдно копiю патенту або лiцензiї, якою дозволяється виробництво та продаж АФI або лiкарського засобу, а також документ, що пiдтверджує чиннiсть патенту в Українi. Заявники подають лист, в якому вказується, що права третьої сторони, захищенi патентом або переданi за лiцензiєю, не порушуються у зв'язку з реєстрацiєю АФI або дiючої речовини. |
||||||||||
2.2. Чи захищена торговельна марка в
Українi?
Якщо так, то наведiть таку iнформацiю:
р Додайте копiї документiв, що передбаченi в пунктi 3.9 цього додатка. |
3. ДОКУМЕНТИ, ЩО ДОДАЮТЬСЯ (позначте необхiдне)
р 3.1. Пiдтвердження оплати (за наявностi).
р 3.2. Доручення для ведення переговорiв/пiдписання документiв вiд iменi заявника (власника реєстрацiйного посвiдчення).
р 3.3. Копiя лiцензiї на виробництво (якщо згiдно iз законодавством країни виробника лiцензiя на виробництво iснує лише в електронному виглядi (наприклад, США), має бути надана роздрукiвка iз посиланням на вiдповiдний офiцiйний сайт, засвiдчена пiдписом/печаткою заявника (для фiзичної особи за наявностi) або iншого дозвiльного документа на виробництво заявленої лiкарської форми у країнi виробника. Копiя повинна бути засвiдчена печаткою заявника (для фiзичної особи за наявностi)/представника заявника в Українi.
р 3.4. Проект технологiчного регламенту або вiдомостi про технологiю виробництва (технологiчна схема виробництва iз зазначенням усiх дiльниць виробництва, задiяних у процесi виробництва АФI або дiючої речовини (уключаючи дiльницi для вiдбору зразкiв i контролю якостi). Копiя повинна бути засвiдчена печаткою заявника (для фiзичної особи - за наявностi)/представника заявника в Українi.
р 3.5. Копiя сертифiката вiдповiдностi Європейськiй фармакопеї (за наявностi).
р 3.6. Сертифiкат вiдповiдностi Європейськiй фармакопеї щодо губчатої енцефалопатiї або документ, виданий уповноваженими органами ветеринарного нагляду країни походження сировини, щодо реєстрацiї в країнi (за результатами клiнiчного та лабораторного контролю) випадкiв губчатої енцефалопатiї.
р 3.7. Копiя письмового дозволу, виданого уповноваженим органом країни виробника, на навмисне вивiльнення ГМО у навколишнє середовище при використаннi з дослiдницькою метою.
р 3.8. Копiї патентiв на винахiд, корисну модель або промисловий зразок, дiя яких розповсюджується на Україну.
р 3.9. Копiї документiв щодо захисту торговельної марки в Українi.
р 3.10. Копiї реєстрацiйного(их) посвiдчення(нь) та вкладки до них (у разi перереєстрацiї).
| Додаток 4 до Порядку проведення експертизи реєстрацiйних матерiалiв на лiкарськi засоби, що подаються на державну реєстрацiю (перереєстрацiю), а також експертизи матерiалiв про внесення змiн до реєстрацiйних матерiалiв протягом дiї реєстрацiйного посвiдчення |
Заява
про державну перереєстрацiю лiкарського засобу
| Дата надходження заяви "___" __________ 20___ року |
N ______________________ |
| Цим я подаю заяву на перереєстрацiю. Я заявляю, що якiсть лiкарського засобу, методи виготовлення та контролю регулярно оновлювалися вiдповiдно до процедури внесення змiн з огляду на технiчний i науковий прогрес. Я пiдтверджую, що до звiту про лiкарський засiб не було внесено змiн, крiм тих, якi схвалено МОЗ. Я гарантую
достовiрнiсть та вiдповiдаю за iнформацiю, що
мiститься у наданих матерiалах реєстрацiйного
досьє. Згоден, що у разi ненадання матерiалiв
реєстрацiйного досьє протягом Усi передбаченi збори сплачено/буде сплачено вiдповiдно до вимог законодавства.
М. П. |
|
1. ОСОБЛИВI ПУНКТИ ЗАЯВИ
| 1.1. Пропозицiї щодо рекламування
лiкарських засобiв, що вiдпускаються без рецепта: р рекламування тiльки серед медичних працiвникiв р рекламування для населення та медичних працiвникiв |
1.2. Заявник (власник реєстрацiйного посвiдчення)/контактна особа
1.2.1. Заявник (власник реєстрацiйного
посвiдчення) (для вiтчизняних виробникiв -
українською, для зарубiжних - українською та
англiйською мовами):
|
1.2.2. Представник заявника
(уповноважена особа, що виступає вiд iменi
заявника):
|
| 1.3. Затвердженi змiни або змiни, що
перебувають на розглядi для внесення або
розширення заяви, якi мали мiсце з дати видачi
реєстрацiйного посвiдчення або останнього
продовження дiї Укажiть у хронологiчному порядку список затверджених змiн або змiн, що перебувають на розглядi. Укажiть дату подачi, дату затвердження (якщо затверджено) i короткий опис змiни. 1. Дата подання номер процедури, якщо необхiдно дата затвердження короткий опис змiни 2. Дата подання номер процедури, якщо необхiдно дата затвердження короткий опис змiни 3. Дата подання номер процедури, якщо необхiдно дата затвердження короткий опис змiни Усi iншi змiни мають бути зазначенi в такому самому форматi |
1.4. Виробники (для вiтчизняних виробникiв - українською, для зарубiжних - українською та англiйською мовами)
1.4.1. Виробник(и), що вiдповiдає(ють) за
випуск серiї
1.4.2. Дiльницi, на яких проводиться контроль серiї, якщо вони вiдмiннi вiд визначених у пунктi 1.4.1:
|
| 1.4.3. Виробник(и) лiкарського засобу i
дiльниця(i) виробництва: Включаючи дiльницi виробництва будь-якого розрiджувача/розчинника в окремiй упаковцi/ємностi, що є частиною лiкарського засобу, дiльницi, на яких здiйснюється контроль якостi/контроль у процесi виробництва, а також (за потреби) iмпортерiв
Короткий опис виконуваних функцiй р Додайте копiю лiцензiї на виробництво (пункт 2.2 цього додатка). р Додайте копiю сертифiката вiдповiдностi вимогам належної виробничої практики, передбаченого у пунктi 2.10 цього додатка. 1.4.4. Виробник(и) ДР та виробничi дiльницi Повиннi бути зазначенi всi дiльницi виробництва, задiянi у виробничому процесi кожного джерела дiючої речовини, включаючи дiльницi, на яких проводиться контроль якостi/контроль у процесi виробництва. Для високотехнологiчних (бiотехнологiчних) лiкарських засобiв мають бути включенi усi дiльницi зберiгання головного та робочого банкiв клiтин та дiльницi приготування робочого банку клiтин.
Короткий опис етапiв виробництва, виконуваних на виробничiй дiльницi р Для кожної ДР додайте заяву уповноваженої особи виробника (пункт 2.13 додатка). · Чи видано сертифiкат вiдповiдностi Європейськiй фармакопеї для ДР?
Якщо так: р Додайте копiю сертифiката вiдповiдностi (пункт 2.6 цього додатка). · Чи є матерiали реєстрацiйного досьє (мастер-файл) на ДР, посилання на якi буде використовуватися у досьє, оригiнальними?
Якщо так: р Додайте лист(и) виробника ДР, що дає(ють) право використання матерiалiв реєстрацiйного досьє (мастер-файл) на ДР (пункт 2.6 цього додатка). р Додайте копiю письмового зобов'язання вiд виробника ДР iнформувати заявника про змiни у виробничому процесi або специфiкацiях (пункт 2.7 цього додатка). · Чи видано сертифiкат на загальний файл на вакцинний антиген (далi - ВАЗФ), що використовується у данiй заявi, або на нього подано заявку?
Якщо так: р Додайте копiю сертифiката на ВАЗФ (пункт 2.8 цього додатка). |
1.5. Якiсний та кiлькiсний склад
| 1.5.1. Якiсний та кiлькiсний склад
лiкарського засобу (ДР та допомiжнi речовини): Необхiдно вказати, на яку кiлькiсть розрахований склад (наприклад, 1 капсула) Перерахуйте дiючi речовини окремо вiд допомiжних речовин:
____________ Iнформацiю про надлишкову кiлькiсть не слiд указувати в колонках щодо складу, а необхiдно викласти нижче:
|
| 1.5.2. Перелiк матерiалiв тваринного
та/або людського походження, що входять до складу
або використовуються у процесi виробництва
лiкарського засобу ВIДСУТНI р
Якщо є сертифiкат вiдповiдностi ЄФ5 щодо ГЕ4 або документ, виданий уповноваженими органами ветеринарного нагляду країни походження сировини, щодо реєстрацiї в країнi (за результатами клiнiчного та лабораторного контролю) випадкiв ГЕ4, вiн повинен бути доданий у пунктi 2.11 цього додатка. ____________ |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||
1.5.3. Чи видано сертифiкат на матерiали
реєстрацiйного досьє (мастер-файл) на плазму (далi
- ПМФ), що використовується у цiй заявi, або на
нього подано заявку?
Якщо так: р Додайте копiю сертифiката (пункт 2.9 цього додатка). |
1.5.4. Чи мiстить або складається
лiкарський засiб з генетично модифiкованих
органiзмiв (ГМО)?
Якщо так, то чи вiдповiдає лiкарський засiб встановленим вимогам? Зробiть необхiдне посилання
р Додайте копiю письмового дозволу, виданого уповноваженим органом країни виробника, на навмисне вивiльнення ГМО у навколишнє середовище при використаннi з дослiдницькою метою (пункт 2.12 цього додатка). |
2. ДОКУМЕНТИ, ЩО ДОДАЮТЬСЯ (позначте необхiдне):
р 2.1. Доручення для ведення переговорiв/пiдписання документiв вiд iменi заявника (власника реєстрацiйного посвiдчення).
р 2.2. Копiя лiцензiї на виробництво (якщо згiдно iз законодавством країни виробника лiцензiя на виробництво iснує лише в електронному виглядi (наприклад, США), має бути надана роздрукiвка iз посиланням на вiдповiдний офiцiйний сайт, засвiдчена пiдписом/печаткою заявника) або iншого дозвiльного документа на виробництво заявленої лiкарської форми у країнi виробника. Копiя повинна бути засвiдчена печаткою заявника/представника заявника в Українi.
р 2.3. Копiї патентiв на винахiд, корисну модель або промисловий зразок, дiя яких розповсюджується на Україну.
р 2.4. Копiї документiв щодо захисту торговельної марки в Українi.
р 2.5. Копiя реєстрацiйного(их) посвiдчення(ь) в Українi разом iз вкладками до реєстрацiйного(их) посвiдчення(ь) та копiя реєстрацiйного посвiдчення або iншого дозвiльного документа (Marketing Authorization, Certificate of a Pharmaceutical Product, Free Sale Certificate тощо), виданого уповноваженим органом країни власника реєстрацiйного посвiдчення (заявника) та/або виробника, або iншої країни, регуляторний орган якої керується високими стандартами якостi, що вiдповiдають стандартам, рекомендованим ВООЗ, на ринку якої розмiшено лiкарський засiб, iз зазначенням назви документа та назви компетентного органу, що його видав, дати видачi i сторiнки, пiдписаної уповноваженою особою компетентного органу. Копiя має бути засвiдчена печаткою заявника/представника заявника в Українi. Зазначити перелiк країн, де даний лiкарський засiб зареєстровано/перереєстровано.
р 2.6. Письмова згода на використання матерiалiв реєстрацiйного досьє на дiючу речовину (у довiльнiй формi) вiд її виробника або копiя сертифiката вiдповiдностi Європейськiй фармакопеї.
р 2.7. Копiя письмового зобов'язання виробника дiючої речовини (у довiльнiй формi) iнформувати заявника про змiни у виробничому процесi або специфiкацiях.
р 2.8. Копiя сертифiката на ВАЗФ.
р 2.9. Копiя сертифiката на ПМФ.
р 2.10. Засвiдчена копiя документа, що пiдтверджує вiдповiднiсть виробництва лiкарського засобу вимогам GMP, виданого Держлiкслужбою вiдповiдно до вимог наказу Мiнiстерства охорони здоров'я України вiд 27 грудня 2012 року N 1130 "Про затвердження Порядку проведення пiдтвердження вiдповiдностi умов виробництва лiкарських засобiв вимогам належної виробничої практики", зареєстрованого у Мiнiстерствi юстицiї України 21 сiчня 2013 року за N 133/22665, або гарантiйного листа заявника щодо надання такого документа протягом строку проведення спецiалiзованої експертизи. За необхiдностi - висновки щодо iнших iнспекцiй, якi проводилися. Копiї документiв мають бути засвiдченi печаткою заявника/представника заявника в Українi.
р 2.11. Сертифiкат вiдповiдностi Європейськiй фармакопеї щодо губчатої енцефалопатiї або документ, виданий уповноваженими органами ветеринарного нагляду країни походження сировини, щодо реєстрацiї в країнi (за результатами клiнiчного та лабораторного контролю) випадкiв губчатої енцефалопатiї.
р 2.12. Копiя письмового дозволу, виданого уповноваженим органом країни виробника, на навмисне вивiльнення ГМО у навколишнє середовище при використаннi з дослiдницькою метою.
р 2.13. Для кожної дiючої речовини дiльниць виробництва, задiяних у виробничому процесi, включаючи дiльницi, на яких проводиться контроль якостi/контроль у процесi виробництва та випуск серiй, заява уповноваженої особи щодо вiдповiдностi виробництва вимогам належної виробничої практики та керiвництвам з належної виробничої практики щодо вихiдних матерiалiв.
| Додаток 5 до Порядку проведення експертизи реєстрацiйних матерiалiв на лiкарськi засоби, що подаються на державну реєстрацiю (перереєстрацiю), а також експертизи матерiалiв про внесення змiн до реєстрацiйних матерiалiв протягом дiї реєстрацiйного посвiдчення |
СТРУКТУРА РЕЄСТРАЦIЙНОГО ДОСЬЄ
(формат загального технiчного документа)
Повне реєстрацiйне досьє складається з п'яти Модулiв:
Модуль 1. АДМIНIСТРАТИВНА IНФОРМАЦIЯ
1.1. Змiст.
1.2. Форма заяви.
1.3. Коротка характеристика лiкарського засобу, маркування та iнструкцiя для медичного застосування:
1.3.1. Копiя короткої характеристики лiкарського засобу/iнструкцiї про застосування лiкарського засобу (iнструкцiї для медичного застосування), затвердженої вiдповiдно до нормативних вимог країни заявника/виробника або iнших країн, регуляторнi органи яких керуються високими стандартами до якостi, що вiдповiдають стандартам, рекомендованим ВООЗ.
1.3.2. Маркування.
1.3.3. Iнструкцiя для медичного застосування (на паперовому та електронному носiях).
1.3.4. Коротка характеристика лiкарського засобу.
1.4. Iнформацiя про незалежних експертiв:
1.4.1. Iнформацiя про експерта з якостi.
1.4.2. Iнформацiя про експерта з доклiнiчних даних.
1.4.3. Iнформацiя про експерта з клiнiчних даних.
1.5. Спецiальнi вимоги до рiзних типiв заяв.
Додаток до Модуля 1. Оцiнка небезпеки для довкiлля.
Модуль 2. РЕЗЮМЕ ЗАГАЛЬНОГО ТЕХНIЧНОГО ДОКУМЕНТА
2.1. Змiст Модулiв 2 - 5.
2.2. Вступ.
2.3. Загальне резюме з якостi.
2.4. Огляд доклiнiчних даних.
2.5. Огляд клiнiчних даних.
2.6. Резюме доклiнiчних даних:
2.6.1. Резюме фармакологiчних даних у текстовому форматi.
2.6.2. Резюме фармакологiчних даних у виглядi таблиць.
2.6.3. Резюме фармакокiнетичних даних у текстовому форматi.
2.6.4. Резюме фармакокiнетичних даних у виглядi таблиць.
2.6.5. Резюме токсикологiчних даних у текстовому форматi.
2.6.6. Резюме токсикологiчних даних у виглядi таблиць.
2.7. Резюме клiнiчних даних:
2.7.1. Резюме бiофармацевтичних дослiджень та пов'язаних з ними аналiтичних методiв.
2.7.2. Резюме дослiджень з клiнiчної фармакологiї.
2.7.3. Резюме з клiнiчної ефективностi.
2.7.4. Резюме з клiнiчної безпеки.
2.7.5. Посилання на джерела лiтератури.
2.7.6. Короткi огляди iндивiдуальних дослiджень.
Модуль 3. ЯКIСТЬ.
ХIМIЧНА, ФАРМАЦЕВТИЧНА ТА БIОЛОГIЧНА IНФОРМАЦIЯ ПРО
ЛIКАРСЬКI ЗАСОБИ, ЩО МIСТЯТЬ ХIМIЧНI ТА/АБО
БIОЛОГIЧНI ДIЮЧI РЕЧОВИНИ
3.1. Змiст.
3.2. Основнi данi.
3.2.S. Дiюча(i) речовина(и).
3.2.S.1. Загальна iнформацiя:
3.2.S.1.1. Назва.
3.2.S.1.2. Структура.
3.2.S.1.3. Загальнi властивостi.
3.2.S.2. Процес виробництва дiючої(их) речовини(н):
3.2.S.2.1. Виробник(и).
3.2.S.2.2. Опис виробничого процесу та його контролю.
3.2.S.2.3. Контроль матерiалiв.
3.2.S.2.4. Контроль критичних стадiй i промiжної продукцiї.
3.2.S.2.5. Валiдацiя процесу та/або його оцiнка.
3.2.S.2.6. Розробка виробничого процесу.
3.2.S.3. Опис характеристик дiючої(их) речовини(н):
3.2.S.3.1. Доказ структури та iншi характеристики.
3.2.S.3.2. Домiшки.
3.2.S.4. Контроль дiючої(их) речовини(н):
3.2.S.4.1. Специфiкацiя.
3.2.S.4.2. Аналiтичнi методики.
3.2.S.4.3. Валiдацiя аналiтичних методик.
3.2.S.4.4. Аналiзи серiй.
3.2.S.4.5. Обґрунтування специфiкацiї.
3.2.S.5. Стандартнi зразки або препарати.
3.2.S.6. Система упаковка/укупорка.
3.2.S.7. Стабiльнiсть:
3.2.S.7.1. Резюме щодо стабiльностi та висновки.
3.2.S.7.2. Протокол пiсляреєстрацiйного вивчення стабiльностi та зобов'язання щодо стабiльностi.
3.2.S.7.3. Данi про стабiльнiсть.
3.2.P. Готовий лiкарський засiб:
3.2.P.1. Опис i склад лiкарського засобу.
3.2.P.2. Фармацевтична розробка:
3.2.P.2.1. Компоненти лiкарського засобу.
3.2.P.2.1.1. Дiюча(i) речовина(и).
3.2.P.2.1.2. Допомiжнi речовини.
3.2.P.2.2. Лiкарський засiб.
3.2.P.2.2.1. Розробка складу.
3.2.P.2.2.2. Надлишки.
3.2.P.2.2.3. Фiзико-хiмiчнi та бiологiчнi властивостi.
3.2.P.2.3. Розробка виробничого процесу.
3.2.P.2.4. Система упаковка/укупорка.
3.2.P.2.5. Мiкробiологiчнi характеристики.
3.2.P.2.6. Сумiснiсть.
3.2.P.3. Процес виробництва лiкарського засобу:
3.2.P.3.1. Виробник(и).
3.2.P.3.2. Склад на серiю.
3.2.P.3.3. Опис виробничого процесу та контролю процесу.
3.2.P.3.4. Контроль критичних стадiй i промiжної продукцiї.
3.2.P.3.5. Валiдацiя процесу та/або його оцiнка.
3.2.P.4. Контроль допомiжних речовин:
3.2.P.4.1. Специфiкацiї.
3.2.P.4.2. Аналiтичнi методики.
3.2.P.4.3. Валiдацiя аналiтичних методик.
3.2.P.4.4. Обґрунтування специфiкацiй.
3.2.P.4.5. Допомiжнi речовини людського або тваринного походження.
3.2.P.4.6. Новi допомiжнi речовини.
3.2.P.5. Контроль лiкарського засобу:
3.2.P.5.1. Специфiкацiя(ї).
3.2.P.5.2. Аналiтичнi методики.
3.2.P.5.3. Валiдацiя аналiтичних методик.
3.2.P.5.4. Аналiзи серiй.
3.2.P.5.5. Характеристика домiшок.
3.2.P.5.6. Обґрунтування специфiкацiї(й).
3.2.P.6. Стандартнi зразки та препарати.
3.2.P.7. Система упаковка/укупорка.
3.2.P.8. Стабiльнiсть:
3.2.P.8.1. Резюме щодо стабiльностi та висновки.
3.2.P.8.2. Протокол пiсляреєстрацiйного вивчення стабiльностi та зобов'язання щодо стабiльностi.
3.2.P.8.3. Данi про стабiльнiсть.
Додаток - iнформацiя щодо:
Примiщення та обладнання.
Оцiнка безпеки щодо стороннiх агентiв.
Новi допомiжнi речовини.
Додаткова iнформацiя.
3.3. Посилання на джерела лiтератури.
Модуль 4. ЗВIТИ ПРО ДОКЛIНIЧНI ДОСЛIДЖЕННЯ
4.1. Змiст.
4.2. Звiти про дослiдження.
4.2.1. Фармакологiя:
4.2.1.1. Первинна фармакодинамiка.
4.2.1.2. Вторинна фармакодинамiка.
4.2.1.3. Фармакологiя безпеки.
4.2.1.4. Фармакодинамiчнi взаємодiї.
4.2.2. Фармакокiнетика:
4.2.2.1. Аналiтичнi методи та звiти щодо їх валiдацiї.
4.2.2.2. Усмоктування.
4.2.2.3. Розподiл.
4.2.2.4. Метаболiзм.
4.2.2.5. Виведення.
4.2.2.6. Фармакокiнетичнi взаємодiї (доклiнiчнi).
4.2.2.7. Iншi фармакокiнетичнi дослiдження.
4.2.3. Токсикологiя:
4.2.3.1. Токсичнiсть при одноразовому введеннi.
4.2.3.2. Токсичнiсть при повторних введеннях.
4.2.3.3. Генотоксичнiсть.
4.2.3.4. Канцерогеннiсть.
4.2.3.5. Репродуктивна токсичнiсть та токсичний вплив на розвиток потомства.
4.2.3.6. Мiсцева переносимiсть.
4.2.3.7. Додатковi дослiдження токсичностi.
4.3. Посилання на джерела лiтератури.
Модуль 5. ЗВIТИ ПРО КЛIНIЧНI ВИПРОБУВАННЯ
5.1. Змiст.
5.2. Перелiк усiх клiнiчних випробувань у виглядi таблиць.
5.3. Звiти про клiнiчнi випробування:
5.3.1. Звiти про бiофармацевтичнi дослiдження.
5.3.2. Звiти, що стосуються дослiдження фармакокiнетики при використаннi бiоматерiалiв людського походження.
5.3.3. Звiти про фармакокiнетичнi дослiдження у людини.
5.3.4. Звiти про фармакодинамiчнi дослiдження у людини.
5.3.5. Звiти про дослiдження ефективностi та безпеки.
5.3.5.1. Звiти про контрольованi клiнiчнi дослiдження щодо пiдтвердження заявлених показань для застосування.
5.3.5.2. Звiти про неконтрольованi клiнiчнi дослiдження, звiти про аналiзи даних за кiлькома дослiдженнями i звiти про iншi клiнiчнi дослiдження.
5.3.6. Звiти про пiсляреєстрацiйний досвiд застосування.
5.3.7. Зразки iндивiдуальних реєстрацiйних форм та iндивiдуальнi списки пацiєнтiв.
5.4. Посилання на джерела лiтератури.
| Додаток 6 до Порядку проведення експертизи реєстрацiйних матерiалiв на лiкарськi засоби, що подаються на державну реєстрацiю (перереєстрацiю), а також експертизи матерiалiв про внесення змiн до реєстрацiйних матерiалiв протягом дiї реєстрацiйного посвiдчення |
Перелiк документiв для проведення експертизи матерiалiв реєстрацiйного досьє для державної реєстрацiї АФI або дiючої речовини
1. Адмiнiстративнi данi.
2. Змiст реєстрацiйного досьє.
3. Форма заяви.
4. Копiя лiцензiї на виробництво (якщо згiдно iз законодавством країни виробника лiцензiя на виробництво iснує лише в електронному виглядi (наприклад, США), має бути надана роздрукiвка iз посиланням на вiдповiдний офiцiйний сайт, засвiдчена пiдписом/печаткою заявника) або iншого дозвiльного документа на виробництво заявленої АФI або дiючої речовини у країнi виробника. Копiя повинна бути засвiдчена печаткою заявника/представника заявника в Українi.
5. Сертифiкати якостi на три промисловi серiї АФI або дiючої речовини або один сертифiкат на одну промислову серiю у супроводi зобов'язання надати сертифiкати на двi iншi серiї, як тiльки вони стануть доступними (усi сертифiкати повиннi подаватися за кожним заявленим мiсцем виробництва).
6. Пропозицiї до маркування.
7. Загальна iнформацiя.
8. Виробництво:
8.1. Виробник(и).
8.2. Опис виробничого процесу та його контролю.
8.3. Контроль матерiалiв.
8.4. Контроль критичних стадiй i промiжної продукцiї.
8.5. Валiдацiя процесу та/або його оцiнка.
9. Опис характеристик:
9.1. Доказ структури та iншi характеристики.
9.2. Домiшки.
10. Контроль:
10.1. Специфiкацiя.
10.2. Аналiтичнi методики.
10.3. Валiдацiя аналiтичних методик.
10.4. Обґрунтування специфiкацiї.
11. Система упаковки/укупорки.
12. Стабiльнiсть:
12.1. Протокол пiсляреєстрацiйного вивчення стабiльностi та зобов'язання щодо стабiльностi (за необхiдностi).
12.2. Данi про стабiльнiсть (за необхiдностi).
Для апробацiї методiв аналiзу якостi АФI або дiючої речовини можуть бути запитанi зразки, еталоннi зразки АФI або дiючої речовини iз сертифiкатами якостi на серiю, включаючи дату виробництва, термiни та умови зберiгання.
| Додаток 7 до Порядку проведення експертизи реєстрацiйних матерiалiв на лiкарськi засоби, що подаються на державну реєстрацiю (перереєстрацiю), а також експертизи матерiалiв про внесення змiн до реєстрацiйних матерiалiв протягом дiї реєстрацiйного посвiдчення |
ПЕРЕЛIК ДОКУМЕНТIВ
для проведення експертизи матерiалiв для
державної перереєстрацiї лiкарського засобу
1. Супровiдний лист.
2. Повний змiст.
3. Заява про державну перереєстрацiю лiкарського засобу (додаток 4).
4. Iнформацiя про уповноважених квалiфiкованих осiб заявника в Українi для здiйснення фармаконагляду.
5. Копiя лiцензiї на виробництво (якщо згiдно iз законодавством країни виробника лiцензiя на виробництво iснує лише в електронному виглядi (наприклад, США), має бути надана роздрукiвка iз посиланням на вiдповiдний офiцiйний сайт, засвiдчена пiдписом/печаткою заявника) або iншого дозвiльного документа на виробництво заявленої лiкарської форми у країнi виробника.
6. Засвiдчена копiя документа, що пiдтверджує вiдповiднiсть виробництва лiкарського засобу вимогам GMP, виданого Держлiкслужбою вiдповiдно до вимог наказу Мiнiстерства охорони здоров'я України вiд 27 грудня 2012 року N 1130 "Про затвердження Порядку проведення пiдтвердження вiдповiдностi умов виробництва лiкарських засобiв вимогам належної виробничої практики", зареєстрованого у Мiнiстерствi юстицiї України 21 сiчня 2013 року за N 133/22665, або гарантiйний лист заявника щодо надання такого документа протягом строку проведення спецiалiзованої експертизи.
7. Копiя реєстрацiйного(их) посвiдчення(ь) в Українi разом iз вкладками до реєстрацiйного(их) посвiдчення(ь) та копiя реєстрацiйного посвiдчення або iншого дозвiльного документа (Marketing Authorization, Certificate of a Pharmaceutical Product, Free Sale Certificate тощо), виданого уповноваженим органом країни власника реєстрацiйного посвiдчення (заявника) та/або виробника, або iншої країни, регуляторний орган якої керується високими стандартами якостi, що вiдповiдають стандартам, рекомендованим ВООЗ, на ринку якої розмiщено лiкарський засiб, iз зазначенням назви документа та назви компетентного органу, що його видав, дати видачi i сторiнки, пiдписаної уповноваженою особою компетентного органу. Копiя має бути засвiдчена печаткою заявника/представника заявника в Українi. Зазначити перелiк країн, де даний лiкарський засiб зареєстровано/перереєстровано.
8. Перелiк одержаних за останнi 5 рокiв в Українi рекламацiй на лiкарський засiб (у хронологiчному порядку) з докладним аналiзом причин, що стали пiдставами для видачi рекламацiй, та заходiв, вжитих заявником для запобiгання їх у майбутньому.
9. Перелiк гарантiй заявника та зобов'язань, наданих пiсля реєстрацiї/перереєстрацiї (у хронологiчному порядку) iз зазначенням характеру, стану виконання, дати подання та дати, коли проблема була вирiшена (рекомендацiї щодо пiсляреєстрацiйних дослiджень, усунення будь-яких недолiкiв, що надавалися контролюючими та iншими органами тощо).
10. Оновлена коротка характеристика лiкарського засобу/iнструкцiя для медичного застосування лiкарського засобу, затвердженi в країнi виробника/заявника; дiючi в Українi iнструкцiя для медичного застосування лiкарського засобу та коротка характеристика лiкарського засобу (за наявностi); проект оновленої iнструкцiї для медичного застосування лiкарського засобу (на паперовому та електронному носiях).
11. Дiючi в Українi методи контролю якостi готового лiкарського засобу, змiни до них (за наявностi) та оновленi методи контролю якостi готового лiкарського засобу.
12. Дiюча версiя частини II або Модуля 3 реєстрацiйного досьє (залежно вiд обраного заявником формату матерiалiв реєстрацiйного досьє).
13. Звiт за результатами пiсляреєстрацiйного нагляду (регулярно оновлюваний звiт з безпеки) та/або узагальнюючий звiт.
14. Зведенi данi виробника/заявника про стан безпеки медичного застосування лiкарського засобу в Українi за перiод дiї останнього реєстрацiйного посвiдчення згiдно з формою, передбаченою вимогами Порядку здiйснення нагляду за побiчними реакцiями лiкарських засобiв, дозволених до медичного застосування, затвердженого наказом МОЗ вiд 27 грудня 2006 року N 898, зареєстрованого у Мiнiстерствi юстицiї України 29 сiчня 2007 року за N 73/13340.
| Примiтки: | 1. При перереєстрацiї лiкарських засобiв у формi in bulk необхiдно надавати документи, перелiченi у пунктах 1, 2, 3, 5, 6, 7, 8, 9, 11, 12 цього додатка. |
| 2. Для окремих груп медичних iмунобiологiчних препаратiв зведенi данi виробника/заявника про стан безпеки його медичного застосування в Українi за перiод дiї останнього реєстрацiйного посвiдчення мiстять: для вакцин та анатоксинiв - данi монiторингу за побiчною дiєю та про iмунологiчну, епiдемiологiчну ефективнiсть; для алергену туберкульозного - данi щодо побiчної дiї та вiдсутностi специфiчної ефективностi. |
| Додаток 8 до Порядку проведення експертизи реєстрацiйних матерiалiв на лiкарськi засоби, що подаються на державну реєстрацiю (перереєстрацiю), а також експертизи матерiалiв про внесення змiн до реєстрацiйних матерiалiв протягом дiї реєстрацiйного посвiдчення |
ПЕРЕЛIК ДОКУМЕНТIВ
для проведення експертизи матерiалiв для
державної перереєстрацiї традицiйних лiкарських
засобiв та лiкарських засобiв, що виробляються
згiдно iз затвердженими прописами
1. Заява про державну реєстрацiю (перереєстрацiю) лiкарського засобу згiдно з додатком 2 до Порядку.
2. Iнформацiя про уповноважених квалiфiкованих осiб заявника для здiйснення фармаконагляду в Українi.
3. Перелiк одержаних за останнi 5 рокiв в Українi рекламацiй на лiкарський засiб (у хронологiчному порядку) з докладним аналiзом причин, що стали пiдставами для видачi рекламацiй, та заходiв, вжитих заявником для запобiгання їм у майбутньому.
4. Копiя лiцензiї на виробництво (якщо згiдно iз законодавством країни виробника лiцензiя на виробництво iснує лише в електронному виглядi (наприклад, США), має бути надана роздрукiвка iз посиланням на вiдповiдний офiцiйний сайт, засвiдчена пiдписом/печаткою заявника) або iншого дозвiльного документа на виробництво заявленої лiкарської форми у країнi виробника.
5. Засвiдчена копiя документа, що пiдтверджує вiдповiднiсть виробництва лiкарського засобу вимогам GMP, виданого Держлiкслужбою вiдповiдно до вимог наказу Мiнiстерства охорони здоров'я України вiд 27 грудня 2012 року N 1130 "Про затвердження Порядку проведення пiдтвердження вiдповiдностi умов виробництва лiкарських засобiв вимогам належної виробничої практики", зареєстрованого у Мiнiстерствi юстицiї України 21 сiчня 2013 року за N 133/22665, або гарантiйний лист заявника щодо надання такого документа протягом строку проведення спецiалiзованої експертизи.
6. Копiя реєстрацiйного(их) посвiдчення(ь) в Українi разом iз вкладками до реєстрацiйного(их) посвiдчення(ь) та копiя реєстрацiйного посвiдчення або iншого дозвiльного документа (Marketing Authorization, Certificate of a Pharmaceutical Product, Free Sale Certificate тощо), виданого уповноваженим органом країни власника реєстрацiйного посвiдчення (заявника) та/або виробника, або iншої країни, регуляторний орган якої керується високими стандартами якостi, що вiдповiдають стандартам, рекомендованим ВООЗ, на ринку якої розмiщено лiкарський засiб, iз зазначенням назви документа та назви компетентного органу, що його видав, дати видачi i сторiнки, пiдписаної уповноваженою особою компетентного органу. Копiя має бути засвiдчена печаткою заявника/представника заявника в Українi. Зазначити перелiк країн, де даний лiкарський засiб зареєстровано/перереєстровано.
7. Проект iнструкцiї для медичного застосування лiкарського засобу.
8. Дiючi в Українi методи контролю якостi готового лiкарського засобу, змiни до них (за наявностi) та оновленi методи контролю якостi готового лiкарського засобу.
9. Вiдомостi про технологiю виробництва (при перереєстрацiї традицiйних лiкарських засобiв).
10. Лист-пiдтвердження стосовно того, що склад, виробництво та контроль лiкарського засобу вiдповiдають пропису, включеному до Перелiку лiкарських засобiв, що виробляються згiдно iз затвердженими прописами, затвердженого наказом Мiнiстерства охорони здоров'я України вiд 11 червня 2009 року N 423.
| Додаток 9 до Порядку проведення експертизи реєстрацiйних матерiалiв на лiкарськi засоби, що подаються на державну реєстрацiю (перереєстрацiю), а також експертизи матерiалiв про внесення змiн до реєстрацiйних матерiалiв протягом дiї реєстрацiйного посвiдчення |
СТРУКТУРА РЕЄСТРАЦIЙНОГО ДОСЬЄ
(у форматi чотирьох частин)
Повне реєстрацiйне досьє складається з чотирьох частин:
Частина I. РЕЗЮМЕ ДОСЬЄ
I. A. Адмiнiстративнi данi.
Змiст реєстрацiйного досьє.
Форма заяви.
I. B. Коротка характеристика лiкарського засобу, iнструкцiя для медичного застосування лiкарського засобу:
I. B. 1. Копiя короткої характеристики лiкарського засобу/iнструкцiї для медичного застосування, затвердженої вiдповiдно до нормативних вимог країни заявника/виробника або iнших країн, регуляторнi органи яких застосовують високi стандарти до якостi, що вiдповiдають стандартам, рекомендованим ВООЗ.
I. B. 2. Пропозицiї до маркування на упаковцi (первиннiй та вториннiй (за наявностi)), iнструкцiї для медичного застосування лiкарського засобу (на паперовому та електронному носiях).
I. B. 3. Коротка характеристика лiкарського засобу.
I. C. Звiти незалежних експертiв:
I. C. 1. Звiт експерта щодо хiмiчної, фармацевтичної та бiологiчної документацiї.
I. C. 2. Звiт експерта щодо фармако-токсикологiчної документацiї.
I. C. 3. Звiт експерта щодо клiнiчної документацiї.
Частина II. ХIМIЧНА, ФАРМАЦЕВТИЧНА ТА БIОЛОГIЧНА ДОКУМЕНТАЦIЯ
Змiст.
II. A. Склад:
II. A. 1. Склад лiкарського засобу.
II. A. 2. Упаковка (короткий опис).
II. A. 3. Склад для клiнiчних випробувань.
II. A. 4. Фармацевтична розробка.
II. B. Метод виготовлення (вiдомостi про технологiю виробництва):
II. B. 1. Виробнича формула.
II. B. 2. Виробничий процес.
II. B. 3. Валiдацiя процесу.
II. C. Методи контролю вихiдних матерiалiв*:
II. C. 1. Дiюча речовина*:
II. C. 1.1. Специфiкацiї та стандартнi методики*.
II. C. 1.2. Науковi данi*:
II. C. 1.2.1. Номенклатура*.
II. C. 1.2.2. Опис*.
II. C. 1.2.3. Виробництво*.
II. C. 1.2.4. Контроль якостi в процесi виробництва.
II. C. 1.2.5. Хiмiчна розробка.
II. C. 1.2.6. Домiшки*.
II. C. 1.2.7. Аналiз серiї*.
____________
* Мiнiмальний обсяг вiдомостей, якi
необхiдно надавати у роздiлi II. C. 1.
II. C. 2. Допомiжнi речовини (ексципiєнти):
II. C. 2.1. Специфiкацiї та встановленi методи контролю якостi.
II. C. 2.2. Науковi данi.
II. C. 3. Пакувальний матерiал (внутрiшня/зовнiшня упаковка):
II. C. 3.1. Специфiкацiї та встановленi методи контролю якостi.
II. C. 3.2. Науковi данi.
II. D. Методи контролю якостi промiжних продуктiв (за необхiдностi).
II. E. Методи контролю якостi готового лiкарського засобу:
II. E. 1. Специфiкацiї та встановленi методи контролю якостi:
II. E. 1.1. Специфiкацiї продукту та методи контролю якостi в процесi виробництва, специфiчнi стандарти.
II. E. 1.2. Методи контролю якостi:
II. E. 1.2.1. Методи для iдентифiкацiї та кiлькiсного визначення дiючої речовини(н).
II. E. 1.2.2. Iдентифiкацiя та визначення ексципiєнта(iв).
II. E. 2. Науковi данi:
II. E. 2.1. Валiдацiя аналiтичних методiв та коментарi, стандарти (робочi стандарти).
II. E. 2.2. Аналiз серiї.
II. F. Стабiльнiсть:
II. F. 1. Методики визначення стабiльностi дiючої речовини(н).
II. F. 2. Методики визначення стабiльностi готового лiкарського засобу.
II. G. Бiодоступнiсть/бiоеквiвалентнiсть.
Дайте посилання на вiдповiднi роздiли частини IV, якщо це необхiдно.
II. H. Данi щодо вiрогiдної небезпеки для навколишнього середовища лiкарських засобiв, якi мiстять генетично модифiкованi мiкроорганiзми (ГМО).
II. Q. Iнша iнформацiя.
Частина III. ФАРМАКОЛОГIЧНА ТА ТОКСИКОЛОГIЧНА ДОКУМЕНТАЦIЯ
Змiст.
III. A. Токсичнiсть при однократному введеннi та введеннi повторних доз:
III. A. 1. Дослiдження токсичностi при однократному введеннi.
III. A. 2. Дослiдження токсичностi при повторних введеннях.
III. B. Репродуктивна функцiя (фертильнiсть та загальна характеристика репродуктивної функцiї).
III. C. Данi щодо ембрiотоксичностi та тератогенностi.
III. D. Мутагенний потенцiал.
III. E. Канцерогенний потенцiал.
III. F. Фармакодинамiка:
III. F.1. Фармакодинамiчнi ефекти вiдносно пропонованих показань.
III. F.2. Загальна фармакодинамiка.
III. F.3. Лiкарськi взаємодiї.
III. G. Фармакокiнетика:
III. G.1. Фармакокiнетика при введеннi однократної дози.
III. G.2. Фармакокiнетика при повторних введеннях.
III. G.3. Розподiл в iнтактних та вагiтних тваринах.
III. G.4. Бiотрансформацiя.
III. H. Мiсцева переноснiсть.
III. Q. Iнша iнформацiя (данi щодо алергенностi тощо).
III. R. Експертиза вiрогiдного ризику для навколишнього середовища/екотоксичнiсть (не обумовлена ГМО).
Частина IV. КЛIНIЧНА ДОКУМЕНТАЦIЯ
Змiст.
IV. A. Клiнiчна фармакологiя:
IV. A.1. Фармакодинамiка.
IV. A.2. Фармакокiнетика.
IV. B. Клiнiчний досвiд:
IV. B.1. Клiнiчнi випробування.
IV. B.2. Пiсляреєстрацiйний досвiд.
IV. B.3. Опублiкованi та неопублiкованi данi щодо клiнiчного досвiду.
IV. C. Надання результатiв.
IV. D. Клiнiчна фармакологiя.
IV. E. Бiодоступнiсть/бiоеквiвалентнiсть.
IV. F. Клiнiчна ефективнiсть та безпека.
IV. G. Матерiали реєстрацiйного досьє, якi супроводжують заяву у виняткових випадках.
IV. H. Пiсляреєстрацiйний досвiд.
IV. Q. Iнша iнформацiя.
Якщо окремi частини документацiї не включенi до матерiалiв реєстрацiйного досьє, то слiд у вiдповiдному мiсцi зазначити причину пiд вiдповiдним заголовком.
Для лiкарських засобiв тваринного походження у роздiлi II.C.1 має бути надана така додаткова iнформацiя:
данi щодо виду, вiку, рацiону тварин, вiд яких отримано сировину;
данi про характер (категорiю) тканини, з якої одержується сировина для виробництва лiкарського засобу, з точки зору її небезпеки щодо вмiсту прiонiв;
технологiчна схема обробки сировини iз зазначенням екстрагентiв, температурного режиму тощо;
методи контролю вихiдної сировини, уключаючи методи виявлення прiонiв у кiнцевому продуктi (за потреби).
| Додаток 10 до Порядку проведення експертизи реєстрацiйних матерiалiв на лiкарськi засоби, що подаються на державну реєстрацiю (перереєстрацiю), а також експертизи матерiалiв про внесення змiн до реєстрацiйних матерiалiв протягом дiї реєстрацiйного посвiдчення |
Структура iнструкцiї
для медичного застосування лiкарського
засобу/лiкарського засобу
(медичного iмунобiологiчного препарату)
Назва лiкарського засобу (укр.)
Англiйською (за бажанням виробника)
Склад:
дiюча(i) речовина(и): (МНН або в разi її вiдсутностi скорочена хiмiчна назва)
склад на 1 одиницю лiкарської форми;
допомiжнi речовини:
Лiкарська форма.
Основнi фiзико-хiмiчнi властивостi:
Фармакотерапевтична група. Код АТХ.
Фармакологiчнi властивостi/Iмунологiчнi i бiологiчнi властивостi
Фармакодинамiка.
Фармакокiнетика.
Клiнiчнi характеристики.
Показання.
Протипоказання.
Особливi заходи безпеки (за наявностi).
Взаємодiя з iншими лiкарськими засобами та iншi види взаємодiй.
Особливостi застосування.
Застосування у перiод вагiтностi або годування груддю.
Здатнiсть впливати на швидкiсть реакцiї при керуваннi автотранспортом або iншими механiзмами.
Спосiб застосування та дози.
Дiти.
Передозування.
Побiчнi реакцiї.
Термiн придатностi.
Умови зберiгання.
Несумiснiсть (за наявностi).
Упаковка.
Категорiя вiдпуску.
Виробник/заявник.
Мiсцезнаходження виробника та його адреса мiсця провадження дiяльностi/мiсцезнаходження заявника та/або представника заявника.
Дата останнього перегляду.
| Додаток 11 до Порядку проведення експертизи реєстрацiйних матерiалiв на лiкарськi засоби, що подаються на державну реєстрацiю (перереєстрацiю), а також експертизи матерiалiв про внесення змiн до реєстрацiйних матерiалiв протягом дiї реєстрацiйного посвiдчення |
МАТЕРIАЛИ РЕЄСТРАЦIЙНОГО ДОСЬЄ,
що подаються для експертизи при внесеннi змiн до
реєстрацiйних матерiалiв протягом дiї
реєстрацiйного посвiдчення
| I. АДМIНIСТРАТИВНI ЗМIНИ |
| 1.1. Змiна найменування та/або мiсцезнаходження заявника (власника реєстрацiйного посвiдчення) | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| 1 | 1, 2 | IА |
| Умови |
| 1. Власником реєстрацiйного посвiдчення повинна залишатися одна й та сама юридична особа або фiзична особа - пiдприємець. |
| Документацiя |
| 1. Документ вiдповiдного компетентного
уповноваженого органу, в якому зазначено нове
найменування та/або нове мiсцезнаходження
заявника (власника реєстрацiйного посвiдчення). 2. Оновленi: коротка характеристика лiкарського засобу, iнструкцiя для медичного застосування та пропозицiї до маркування на упаковцi лiкарського засобу (за необхiдностi). |
| 1.2. Змiна торговельної назви лiкарського засобу | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| 1 | 1, 2 | IБ |
| Умови |
| 1. Запропонована назва не порушує права третiх сторiн. |
| Документацiя |
| 1. Обґрунтування змiни назви лiкарського
засобу. 2. Оновленi: коротка характеристика лiкарського засобу, iнструкцiя для медичного застосування та пропозицiї до маркування на упаковцi лiкарського засобу. |
| 1.3. Змiна назви АФI або дiючої речовини | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| 1 | 1, 2 | IА |
| Умови |
| 1. АФI або дiюча речовина повинна залишатися тiєю самою. |
| Документацiя |
| 1. Пiдтвердження рекомендованих ВООЗ
мiжнародних непатентованих назв або копiя їх
перелiку. Для рослинних лiкарських засобiв - заява
про те, що назва вiдповiдає чинним вимогам або
чинному Керiвництву з якостi рослинних лiкарських
засобiв та керiвним принципам для декларування
рослинних субстанцiй та рослинних препаратiв, що
входять до складу (традицiйних) рослинних
лiкарських засобiв. 2. Оновленi: коротка характеристика лiкарського засобу, iнструкцiя для медичного застосування та пропозицiї до маркування на упаковцi лiкарського засобу. |
| 1.4. Змiна найменування та/або мiсцезнаходження виробника (включаючи за необхiдностi мiсце проведення контролю якостi) або постачальника АФI або дiючої речовини/вихiдного матерiалу/реагенту/промiжного продукту, що застосовуються у виробництвi АФI або дiючої речовини (за вiдсутностi сертифiката вiдповiдностi Європейськiй фармакопеї у затвердженому реєстрацiйному досьє) | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| 1 | 1, 2, 3 | IА |
| Умови |
| 1. Виробнича дiльниця та усi виробничi операцiї залишаються незмiнними. |
| Документацiя |
| 1. Документ вiдповiдного уповноваженого
органу, у якому зазначено нове найменування
та/або мiсцезнаходження. 2. Змiни до вiдповiдних роздiлiв матерiалiв реєстрацiйного досьє. 3. У разi змiни у найменуваннi власника мастер-файла на дiючу речовину - поновлений дозвiл власника на користування даними, що мiстяться у мастер-файлi. |
| 1.5. Змiна найменування та/або мiсцезнаходження виробника готового лiкарського засобу, включаючи мiсце проведення контролю якостi | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) виробнича дiльниця випуску серiй | 1 | 1, 2 | IА |
| б) усi iншi дiльницi | 1 | 1, 2 | IА |
| Умови |
| 1. Виробнича дiльниця та всi виробничi операцiї залишаються незмiнними. |
| Документацiя |
| 1. Копiя оновленої лiцензiї на
виробництво (якщо згiдно iз законодавством країни
виробника лiцензiя на виробництво iснує лише в
електронному виглядi (наприклад, США), має бути
надана роздрукiвка iз посиланням на вiдповiдний
офiцiйний сайт, засвiдчена пiдписом/печаткою
заявника) або iншого дозвiльного документа на
виробництво заявленої лiкарської форми у країнi
виробника, у якому зазначено нове найменування
та/або мiсцезнаходження, та засвiдчена копiя
документа, що пiдтверджує вiдповiднiсть
виробництва лiкарського засобу вимогам GMP,
виданого Держлiкслужбою вiдповiдно до вимог
наказу Мiнiстерства охорони здоров'я України вiд 27
грудня 2012 року N 1130 "Про затвердження
Порядку проведення пiдтвердження вiдповiдностi
умов виробництва лiкарських засобiв вимогам
належної виробничої практики",
зареєстрованого у Мiнiстерствi юстицiї України 21
сiчня 2013 року за N 133/22665, або гарантiйний лист
заявника щодо надання такого документа протягом
строку проведення спецiалiзованої експертизи. 2. Змiни до вiдповiдних роздiлiв матерiалiв реєстрацiйного досьє, включаючи оновленi коротку характеристику лiкарського засобу, iнструкцiю для медичного застосування та пропозицiї до маркування на упаковцi лiкарського засобу (за необхiдностi). |
| 1.6. Змiна коду АТХ | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| 1 | 1, 2 | IА |
| Умови |
| 1. Присвоєння нового або змiна ВООЗ коду АТХ. |
| Документацiя |
| 1. Пiдтвердження присвоєння ВООЗ коду
АТХ або копiя перелiку кодiв АТХ. 2. Оновленi: коротка характеристика лiкарського засобу, iнструкцiя для медичного застосування та пропозицiї до маркування на упаковцi лiкарського засобу (за необхiдностi). |
| 1.7. Вилучення виробничої дiльницi (включаючи дiльницi для АФI або дiючої речовини, промiжного продукту або готового лiкарського засобу, дiльницi для проведення пакування, виробника, вiдповiдального за випуск серiй, мiсце проведення контролю серiї) або постачальника вихiдного матерiалу, реагенту або допомiжної речовини (якщо зазначено у реєстрацiйному досьє) | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| 1, 2 | 1, 2 | IА |
| Умови |
| 1. Необхiдно залишити принаймнi одну
затверджену дiльницю/виробника, що виконують
таку саму функцiю, що й вилученi. 2. Вилучення не обумовлено непередбаченими обставинами у виробничому процесi. |
| Документацiя |
| 1. Заява про внесення змiн має чiтко
визначати затвердженого та запропонованого
виробникiв, як зазначено в пунктi 2.5 заяви про
державну реєстрацiю лiкарського засобу, форма
якої наведена у додатку 1 до Порядку. 2. Змiни до вiдповiдних роздiлiв реєстрацiйного досьє, включаючи оновленi: коротка характеристика лiкарського засобу, iнструкцiя для медичного застосування та пропозицiї до маркування на упаковцi лiкарського засобу (за необхiдностi). |
| II. ЗМIНИ ЩОДО ЯКОСТI |
| 2.1. АФI або дiюча речовина |
| 2.1.1. Виробництво |
| 2.1.1.1. Змiна виробника вихiдного/промiжного продукту/реагенту, що використовується у виробничому процесi АФI або дiючої речовини, або змiна виробника (включаючи, де необхiдно, мiсце проведення контролю якостi) АФI або дiючої речовини (за вiдсутностi сертифiката вiдповiдностi Європейськiй фармакопеї у матерiалах реєстрацiйного досьє) | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) запропонована додаткова виробнича дiльниця є дiльницею одного виробника | 1, 2, 3 | 1, 2, 3, 4, 5, 6, 7 | IА |
| б) введення нового виробника АФI або дiючої речовини з наданням матерiалiв реєстрацiйного досьє (мастер-файла) на дiючу речовину | II | ||
| в) запропонований виробник використовує спосiб синтезу, що суттєво вiдрiзняється вiд попереднього, або умови виробництва, якi потенцiйно можуть змiнити важливi характеристики якостi АФI або дiючої речовини, такi як якiсний та/або кiлькiсний профiль домiшок, що потребує квалiфiкацiї, або фiзико-хiмiчнi властивостi АФI або дiючої речовини, що впливають на бiодоступнiсть | II | ||
| г) новий виробник вихiдного продукту, для якого вимагається попередня оцiнка вiрусної безпеки та/або ризику передачi збудникiв губчатої енцефалопатiї | II | ||
| ґ) змiна у АФI або дiючої речовини бiологiчного походження або вихiдному матерiалi/реагентi/ промiжному продуктi, що використовується для виробництва лiкарського засобу бiологiчного походження | II | ||
| д) змiни у методах контролю якостi АФI або дiючої речовини або додавання дiльницi для проведення контролю серiї/випробування | 2, 4 | 1, 5 | IА |
| Умови |
| 1. Для вихiдних матерiалiв та реагентiв
специфiкацiї (включаючи контроль пiд час
виробництва, методи аналiзу всiх матерiалiв)
iдентичнi затвердженим. Для промiжних продуктiв та
АФI або дiючих речовин специфiкацiї (включаючи
контроль пiд час виробництва, методи аналiзу всiх
матерiалiв) спосiб виробництва (включаючи розмiр
серiї) та детальний опис методу синтезу - iдентичнi
затвердженим. 2. АФI або дiюча речовина не є стерильною або речовиною бiологiчного/iмунологiчного походження. 3. Якщо у процесi виробництва використовуються матерiали людського або тваринного походження, виробник не використовує будь-якого нового постачальника, для якого потрiбна оцiнка вiрусної безпеки або вiдповiдностi виробництва Керiвництву з мiнiмiзацiї ризику передачi збудникiв губчатої енцефалопатiї тварин з лiкарськими засобами. 4. Перенесення способу виробництва з попередньої дiльницi до нової було успiшно виконано. |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє (за необхiдностi). 2. Заява вiд власника реєстрацiйного посвiдчення або вiд власника мастер-файла на дiючу речовину (за необхiдностi), що метод синтезу (або у разi лiкарських засобiв рослинного походження за потреби метод приготування, мiсце походження, виробництво рослинної субстанцiї та спосiб виробництва), методи контролю якостi та специфiкацiї на АФI або дiючу речовину та вихiдний матерiал/реагент/промiжний продукт у виробництвi АФI або дiючої речовини не вiдрiзняються вiд затверджених за потреби. 3. Сертифiкат вiдповiдностi Європейськiй фармакопеї щодо губчатої енцефалопатiї для нового вихiдного продукту або, якщо необхiдно, документальне пiдтвердження того, що вихiдний продукт для отримання АФI або дiючої речовини, який має ризик передачi збудникiв губчатої енцефалопатiї, попередньо оцiнений уповноваженим органом країни виробника та продемонстрована його вiдповiднiсть Керiвництву з мiнiмiзацiї ризику передачi збудникiв губчатої енцефалопатiї тварин з лiкарськими засобами (чинне видання). Iнформацiя повинна мiстити: найменування виробника, види та тканини тварин, з яких одержано вихiдний продукт, назву країни походження тваринної сировини, її використання та попереднiй документ. 4. Данi аналiзiв (у виглядi таблиць порiвняння) принаймнi для двох серiй (мiнiмум дослiдно-промислових) АФI або дiючої речовини вiд затвердженого та запропонованого виробникiв/дiльниць. 5. Заява про внесення змiн має мiстити чiтку iнформацiю щодо "затвердженого" та "запропонованого" виробникiв, як зазначено в пунктi 2.5 заяви про державну реєстрацiю лiкарського засобу, форма якої наведена у додатку 1 до Порядку. 6. Заява уповноваженої особи (далi - УО) кожного з виробникiв, перелiчених у заявi, якщо АФI або дiюча речовина використовується як вихiдний матерiал, та заява УО кожного з перелiчених у заявi виробникiв, вiдповiдальних за випуск серiї. У заявах УО необхiдно зазначити, що виробник(и) АФI або дiючої речовини дiє вiдповiдно до настанов та керiвництв з належної виробничої практики щодо вихiдних матерiалiв. Узагальнена заява може бути прийнятною за умов, приведених у примiтцi до пiдпункту 2.2.2.1 цього додатка. 7. Зобов'язання виробника АФI або дiючої речовини iнформувати власника реєстрацiйного посвiдчення про будь-якi змiни у процесi виробництва, специфiкацiях та методах аналiзу АФI або дiючої речовини (за необхiдностi). |
| 2.1.1.2. Змiни в процесi виробництва АФI або дiючої речовини | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) незначна змiна у процесi виробництва АФI або дiючої речовини | 1, 2, 3, 4, 5, 6, 7 | 1, 2, 3 | IА |
| б) значна змiна у процесi виробництва АФI або дiючої речовини, що може мати iстотний вплив на якiсть, безпеку або ефективнiсть лiкарського засобу | II | ||
| в) змiна стосується АФI або дiючої речовини бiологiчного/iмунологiчного походження або використання хiмiчних субстанцiй у виробництвi лiкарського засобу бiологiчного/iмунологiчного походження i не вiдноситься до протоколу виробництва | II | ||
| г) змiна у лiкарському засобi рослинного походження, що стосується однiєї з таких характеристик: джерело походження сировини, спосiб виробництва або виготовлення | II | ||
| ґ) незначна змiна у закритiй частинi матерiалiв реєстрацiйного досьє (мастер-файла) на дiючу речовину | 1, 2, 3, 4 | IБ |
| Умови |
| 1. Не повинно бути жодних змiн у якiсному
та кiлькiсному складi домiшок або змiн
фiзико-хiмiчних властивостей АФI або дiючої
речовини. 2. Шлях синтезу не змiнюється, а саме промiжнi продукти залишаються незмiнними, i не використовуються новi реагенти, каталiзатори або розчинники. Для рослинних лiкарських засобiв - джерело походження сировини, виробництво субстанцiї та спосiб виробництва залишаються незмiнними. 3. Специфiкацiї на АФI або дiючу речовину або промiжнi продукти залишаються незмiнними. 4. Змiна цiлком описана у вiдкритiй для заявника частинi мастер-файла на дiючу речовину (за необхiдностi). 5. АФI або дiюча речовина не є речовиною бiологiчного/iмунологiчного походження. 6. Змiна не стосується джерела походження та способу виробництва рослинного лiкарського засобу. 7. Змiна не стосується закритої частини мастер-файла на дiючу речовину. |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє та затвердженого
мастер-файла на дiючу речовину (за можливостi),
включаючи результати порiвняння узгодженого та
запропонованого виробничого процесу. 2. Данi аналiзiв (у виглядi таблицi порiвняння) принаймнi для двох серiй (мiнiмум дослiдно-промислових) лiкарського засобу (АФI або дiючої речовини), виготовлених вiдповiдно до узгодженого та запропонованого виробничого процесу. 3. Копiя затверджених специфiкацiй на АФI або дiючу речовину. 4. Заява вiд власника реєстрацiйного посвiдчення або власника мастер-файла на дiючу речовину (за необхiдностi), що будь-якi змiни у якiсному та кiлькiсному профiлi домiшок або у фiзико-хiмiчних властивостях - вiдсутнi, метод синтезу не змiнюється, а також не змiнюються специфiкацiї на АФI або дiючу речовину або промiжнi продукти. |
| Примiтка до пiдпункту "б" пiдпункту 2.1.1.2 цього додатка. Для хiмiчних АФI або дiючих речовин це стосується значних змiн у методi синтезу або умовах виробництва, що потенцiйно можуть змiнювати важливi характеристики АФI або дiючої речовини, такi як якiсний та/або кiлькiсний профiль домiшок, що потребує квалiфiкацiї, або фiзико-хiмiчнi властивостi АФI або дiючої речовини, що впливають на бiодоступнiсть. |
| 2.1.1.3. Змiна розмiру серiї (включаючи дiапазони) АФI або дiючої речовини або промiжного продукту | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) збiльшення до 10 разiв порiвняно iз затвердженим розмiром серiї | 1, 2, 3, 4, 6, 7, 8 | 1, 2, 5 | IА |
| б) зменшення обсягу виробництва | 1, 2, 3, 4, 5 | 1, 2, 5 | IА |
| в) змiна розмiру серiї, що потребує доведення подiбностi АФI або дiючої речовини бiологiчного/iмунологiчного походження порiвняно iз затвердженою | II | ||
| г) збiльшення у понад 10 разiв порiвняно iз затвердженим розмiром серiї | 1, 2, 3, 4 | IБ | |
| ґ) розмiр серiї АФI або дiючої речовини бiологiчного/iмунологiчного походження збiльшився/зменшився без змiни параметрiв процесу (наприклад, дублювання лiнiї) | 1, 2, 3, 4 | IБ |
| Умови |
| 1. Змiни у процесi виробництва обумовленi
тiльки збiльшенням або зменшенням обсягу
виробництва, наприклад, використанням
обладнання iншої продуктивностi. 2. Наявнiсть результатiв аналiзу принаймнi двох серiй запропонованого розмiру серiї вiдповiдно до специфiкацiй. 3. АФI або дiюча речовина не є речовиною бiологiчного/iмунологiчного походження. 4. Змiна не повинна впливати на вiдтворюванiсть процесу виробництва. 5. Змiна не повинна бути обумовлена непередбаченими обставинами в процесi виробництва або проблемами, пов'язаними зi стабiльнiстю. 6. Специфiкацiї на АФI або дiючi речовини/промiжнi продукти залишаються незмiнними. 7. АФI або дiюча речовина не є стерильною. 8. Затверджений розмiр серiї не був схвалений при внесеннi змiн типу IА. |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє. 2. Номери серiй запропонованого розмiру серiї, якi проходять випробування. 3. Данi аналiзiв (у виглядi таблицi порiвняння) принаймнi однiєї промислової серiї АФI або дiючої речовини або промiжного продукту для затвердженого та запропонованого розмiрiв серiї (за необхiдностi). Данi про наступнi двi повнi промисловi серiї мають бути наданi на вимогу, а також заява про те, що у разi невiдповiдностi специфiкацiям буде повiдомлено Центр (з вiдповiдною пропозицiєю). 4. Копiя затверджених специфiкацiй на АФI або дiючу речовину (та промiжнi продукти за потреби). 5. Заява вiд власника реєстрацiйного посвiдчення або власника мастер-файла на дiючу речовину про те, що змiни у процесi виробництва обумовленi тiльки збiльшенням або зменшенням обсягу виробництва, наприклад, використанням обладнання iншої продуктивностi, що змiна не впливає на вiдтворюванiсть процесу i не обумовлена непередбаченими обставинами в процесi виробництва або проблемами, пов'язаними зi стабiльнiстю, i що специфiкацiї на АФI або дiючу речовину/промiжнi продукти залишаються незмiнними. |
| 2.1.1.4. Змiни випробувань або допустимих меж, що встановленi у специфiкацiях, у процесi виробництва АФI або дiючої речовини | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) звуження допустимих меж | 1, 2, 3, 4 | 1, 2 | IА |
| б) додавання нового випробування та допустимих меж | 1, 2, 5, 6 | 1, 2, 3, 4, 6 | IА |
| в) вилучення несуттєвого випробування | 1, 2 | 1, 2, 5 | IА |
| г) розширення затверджених допустимих меж для показникiв, якi можуть iстотно вплинути на якiсть АФI або дiючої речовини | II | ||
| ґ) вилучення випробування, що може мати iстотний влив на загальну якiсть АФI або дiючої речовини | II | ||
| д) додавання або замiна випробування за результатами дослiджень з безпеки або якостi | 1, 2, 3, 4, 6 | IБ |
| Умови |
| 1. Змiна не обумовлена попередньою
експертизою, що здiйснювалась для перегляду
показникiв, зазначених у специфiкацiї (наприклад,
проведеною пiд час подання заяви про реєстрацiю
або внесення змiн типу II). 2. Змiна не обумовлена непередбаченими обставинами в процесi виробництва, наприклад, появою нової неквалiфiкованої домiшки; змiною меж загального вмiсту домiшок. 3. Будь-якi змiни не повиннi виходити за допустимi межi затвердженої специфiкацiї. 4. Методи випробувань залишилися незмiнними або такi змiни є незначними. 5. Будь-який новий метод випробувань не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосiб. 6. Новий метод випробувань не належить до бiологiчних/iмунологiчних/iмунохiмiчних методiв або методу, при якому використовується бiологiчний реагент для аналiзу АФI або дiючої речовини бiологiчного походження (за винятком стандартних фармакопейних мiкробiологiчних методiв). |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє. 2. Порiвняльна таблиця щодо затверджених та запропонованих методiв випробувань. 3. Опис нового нефармакопейного методу випробування та данi з його валiдацiї (за необхiдностi). 4. Данi аналiзiв для двох промислових серiй (трьох промислових серiй для АФI або дiючої речовини бiологiчного походження, якщо не обґрунтовано iнше) АФI або дiючої речовини за всiма показниками, що зазначенi у специфiкацiї. 5. Обґрунтування/оцiнка ризику вiд власника реєстрацiйного посвiдчення або власника мастер-файла на дiючу речовину (за необхiдностi), що пiдтверджує незначнiсть змiненого параметра. 6. Обґрунтування вiд власника реєстрацiйного посвiдчення або власника мастер-файла на дiючу речовину (за необхiдностi) введення нового методу випробування або допустимих меж, визначених у специфiкацiях. |
| 2.1.1.5. Змiни у АФI або дiючiй речовинi для сезонних, передпандемiчних або пандемiчних вакцин для профiлактики грипу | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) замiна штаму(iв) у сезонних, передпандемiчних або пандемiчних вакцинах для профiлактики грипу | II |
| 2.1.2. Контроль АФI або дiючої речовини |
| 2.1.2.1. Змiна у параметрах специфiкацiй та/або допустимих меж, визначених у специфiкацiях на АФI або дiючу речовину, або вихiдний/промiжний продукт/реагент, що використовуються у процесi виробництва АФI або дiючої речовини | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) звуження допустимих меж, визначених у специфiкацiї на лiкарськi засоби, що пiдлягають випуску серiї | 1, 2, 3, 4 | 1, 2 | IА |
| б) звуження допустимих меж, визначених у специфiкацiї | 1, 2, 3, 4 | 1, 2 | IА |
| в) доповнення специфiкацiї новим показником якостi та вiдповiдним методом випробування | 1, 2, 5, 6, 7 | 1, 2, 3, 4, 7 | IА |
| г) вилучення незначного показника якостi (наприклад, вилучення застарiлого показника) | 1, 2 | 1, 2, 6 | IА |
| ґ) вилучення параметра специфiкацiї, який може мати суттєвий влив на якiсть АФI або дiючої речовини та/або готового лiкарського засобу | II | ||
| д) розширення допустимих меж, визначених у специфiкацiї на АФI або дiючу речовину | II | ||
| е) розширення допустимих меж, визначених у специфiкацiях на вихiднi матерiали/промiжнi продукти, якi мають iстотний вплив на якiсть АФI або дiючої речовини та/або готового лiкарського засобу | II | ||
| є) доповнення або замiна параметра специфiкацiї за результатами дослiджень з безпеки або якостi (за винятком АФI або дiючої речовини бiологiчного або iмунологiчного походження) | 1, 2, 3, 4, 5, 7 | IБ |
| Умови |
| 1. Змiна не обумовлена попередньою
експертизою, що здiйснювалась для перегляду
показникiв, зазначених у специфiкацiї (наприклад,
проведеною пiд час подання заяви про реєстрацiю
або внесення змiн типу II). 2. Змiна не обумовлена непередбаченими обставинами в процесi виробництва, наприклад, появою нової неквалiфiкованої домiшки; змiною меж загального вмiсту домiшок. 3. Будь-якi змiни не повиннi виходити за допустимi межi затвердженої специфiкацiї. 4. Методи випробувань залишилися незмiнними або такi змiни є незначними. 5. Будь-який новий метод випробувань не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосiб. 6. Новий метод випробувань не належить до бiологiчних/iмунологiчних/iмунохiмiчних методiв або методу, при якому використовується бiологiчний реагент для аналiзу АФI або дiючої речовини бiологiчного походження (за винятком стандартних фармакопейних мiкробiологiчних методiв). 7. Змiна не стосується домiшки, яка має генотоксичну дiю. |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє. 2. Порiвняльна таблиця щодо затверджених та запропонованих специфiкацiй. 3. Опис нового аналiтичного методу та данi з його валiдацiї (за необхiдностi). 4. Результати аналiзу для двох промислових серiй (трьох промислових серiй для АФI або дiючих речовин бiологiчного походження, якщо не обґрунтовано iнше) АФI або дiючої речовини за всiма показниками, що зазначенi у специфiкацiї. 5. Порiвняльнi данi щодо профiлю розчинення готового лiкарського засобу для принаймнi однiєї дослiдно-промислової серiї, що мiстить АФI або дiючу речовину за затвердженою та запропонованою специфiкацiями (за необхiдностi). Для рослинних лiкарських засобiв можуть бути прийнятнi данi щодо розпадання. 6. Обґрунтування/оцiнка ризику вiд власника реєстрацiйного посвiдчення або власника мастер-файла на дiючу речовину (за необхiдностi), що пiдтверджує незначнiсть змiненого параметра. 7. Обґрунтування вiд власника реєстрацiйного посвiдчення або власника мастер-файла на дiючу речовину (за необхiдностi) введення нового методу випробування або допустимих меж, визначених у специфiкацiях. |
| 2.1.2.2. Змiна у методах випробування АФI або дiючої речовини або вихiдного/промiжного продукту/реагенту, що використовується у процесi виробництва АФI або дiючої речовини | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) незначнi змiни у затверджених методах випробування | 1, 2, 3, 4 | 1, 2 | IА |
| б) видалення методу випробування для АФI або дiючої речовини /реагенту/промiжного продукту, якщо альтернативний метод вже затверджений | 7 | 1 | IА |
| в) iншi змiни в методах випробування (включаючи замiну або доповнення) для реагенту, що не спричиняє iстотного впливу на якiсть АФI або дiючої речовини | 1, 2, 3, 5, 6 | 1, 2 | IА |
| г) змiна у бiологiчному/iмубiонологiчному/ iмунохiмiчному методi випробування або методi, у якому використовується бiологiчний реагент для АФI або дiючої речовини бiологiчного походження, наприклад, пептиднi карти, глiк-карти тощо | II | ||
| ґ) iншi змiни у методах випробування (включаючи замiну або доповнення) АФI або дiючої речовини або вихiдного/промiжного продукту | 1, 2 | IБ |
| Умови |
| 1. Дослiдження з валiдацiї, якi були
проведенi вiдповiдно до чинних фармакопейних
вимог або настанов та керiвних принципiв з
валiдацiї, пiдтверджують, що результати аналiзу,
отриманi за затвердженою та запропонованою
методиками, - iдентичнi. 2. Не вiдбулося жодних змiн меж загального вмiсту домiшок, не виявлено нових неквалiфiкованих домiшок. 3. Методи випробувань залишилися незмiнними (наприклад, змiнилась довжина колонки або температура проведення аналiзу, але тип колонки або методика - незмiннi). 4. Новий метод випробувань не належить до бiологiчних/iмунологiчних/iмунохiмiчних методiв або методу, при якому використовується бiологiчний реагент для аналiзу АФI або дiючої речовини бiологiчного походження (за винятком стандартних фармакопейних мiкробiологiчних методiв). 5. Будь-який новий метод випробування не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосiб. 6. АФI або дiюча речовина не є речовиною бiологiчного/iмунологiчного походження. 7. Iснує затверджений метод випробування для показника специфiкацiї i цей метод не затверджений при внесеннi змiни типу IА. |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє, включаючи опис методiв
контролю, звiт про данi з валiдацiї, переглянутi
допустимi межi для домiшок (за необхiдностi). 2. Порiвняльнi данi з валiдацiї або, якщо обґрунтовано, порiвняльнi результати аналiзу, якi пiдтверджують, що затверджена та запропонована методики випробування - iдентичнi. Ця вимога не стосується випадку додавання нової методики випробування. |
| 2.1.3. Система упаковка/укупорка |
| 2.1.3.1. Змiна у безпосереднiй упаковцi АФI або дiючої речовини | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) якiснi та/або кiлькiснi змiни складу | 1, 2, 3 | 1, 2, 3, 4, 6 | IА |
| б) якiснi та/або кiлькiснi змiни складу для стерильних та незаморожених АФI або дiючих речовин бiологiчного/iмунологiчного походження | II | ||
| в) рiдких АФI або дiючих речовин (нестерильних) | 1, 2, 3, 5, 6 | IБ |
| Умови |
| 1. Запропонований пакувальний матерiал
має бути iдентичним затвердженому за вiдповiдними
показниками якостi. 2. Дослiдження стабiльностi розпочато вiдповiдно до настанови щодо випробування стабiльностi або керiвних принципiв IСН щодо випробування стабiльностi (чинне видання) принаймнi для двох дослiдно-промислових або промислових серiй АФI або дiючої речовини i на дату випуску у розпорядженнi заявника були задовiльнi данi зi стабiльностi принаймнi за три мiсяцi. Однак, якщо запропонована упаковка стiйкiша, нiж затверджена, данi щодо стабiльностi за три мiсяцi ще можуть бути недоступнi. При цьому мають бути наданi гарантiї, що дослiдження будуть завершенi, i одразу пiсля завершення дослiджень у разi невiдповiдностi специфiкацiям або наявної ймовiрностi невiдповiдностi специфiкацiям наприкiнцi термiну придатностi/перiоду повторного випробування данi будуть поданi до Центру (iз запропонованими заходами). 3. Не стосується стерильних рiдких АФI або дiючих речовин та АФI або дiючих речовин бiологiчного/iмунологiчного походження. |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє. 2. Вiдповiднi данi щодо нової упаковки (наприклад, порiвняльнi данi про проникнiсть, наприклад, для O2, CO2, вологи), включаючи пiдтвердження того, що пакувальний матерiал вiдповiдає чинним фармакопейним вимогам щодо пакувальних матерiалiв або законодавству ЄС щодо безпеки взаємодiї пластикових пакувальних матерiалiв та упаковок з харчовими продуктами. 3. Пiдтвердження вiдсутностi будь-якої взаємодiї мiж АФI або дiючою речовиною та пакувальним матерiалом (наприклад, вiдсутнє перенесення компонентiв запропонованого пакувального матерiалу до АФI або дiючої речовини та немає жодних втрат компонентiв упаковки), включаючи пiдтвердження того, що пакувальний матерiал вiдповiдає чинним фармакопейним вимогам щодо пакувальних матерiалiв або законодавству ЄС щодо безпеки взаємодiї пластикових пакувальних матерiалiв та упаковок з харчовими продуктами. 4. Пiдтвердження вiд власника реєстрацiйного посвiдчення або власника мастер-файла на дiючу речовину (за необхiдностi), що дослiдження стабiльностi розпочато вiдповiдно до настанови щодо випробування стабiльностi або керiвних принципiв IСН щодо випробування стабiльностi (чинне видання) (iз зазначенням кiлькостi та номерiв серiй АФI або дiючої речовини) та пiдтвердження (за необхiдностi), що на момент внесення змiн заявником було надано мiнiмум, який вимагається, даних щодо стабiльностi та наявнi данi не вказують на будь-яку проблему, пов'язану зi стабiльнiстю. Мають бути наданi гарантiї, що дослiдження будуть завершенi, i одразу пiсля завершення дослiджень у разi невiдповiдностi специфiкацiям або наявної ймовiрностi невiдповiдностi специфiкацiям наприкiнцi термiну придатностi/перiоду повторного випробування данi будуть поданi до Центру (iз запропонованими заходами). 5. Результати дослiджень стабiльностi, проведених згiдно з настановою щодо випробування стабiльностi або керiвних принципiв IСН щодо випробування стабiльностi (чинне видання) за вiдповiдними параметрами принаймнi для двох дослiдно-промислових або промислових серiй АФI або дiючої речовини iз задовiльними показниками щодо стабiльностi принаймнi за три мiсяцi. При цьому мають бути наданi гарантiї, що дослiдження будуть завершенi, i одразу пiсля завершення дослiджень у разi невiдповiдностi специфiкацiям або наявної ймовiрностi невiдповiдностi специфiкацiям наприкiнцi термiну придатностi/перiоду повторного випробування данi будуть поданi до Центру (iз запропонованими заходами). 6. Порiвняльна таблиця вимог затвердженої та запропонованої специфiкацiй (за необхiдностi). |
| 2.1.3.2. Змiни у параметрах специфiкацiй та/або допустимих меж, зазначених у специфiкацiях, для первинної упаковки АФI або дiючої речовини | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) звуження допустимих меж, зазначених у специфiкацiях | 1, 2, 3, 4 | 1, 2 | IА |
| б) доповнення специфiкацiї новим показником та вiдповiдним методом випробування | 1, 2, 5 | 1, 2, 3, 4, 6 | IА |
| в) вилучення незначного показника (наприклад, вилучення застарiлого показника) | 1, 2 | 1, 2, 5 | IА |
| г) доповнення або замiна показника за результатами дослiджень з безпеки або якостi | 1, 2, 3, 4, 6 | IБ |
| Умови |
| 1. Змiна не обумовлена попередньою
експертизою, що здiйснювалась для перегляду
показникiв, зазначених у специфiкацiї (наприклад,
проведеною пiд час подання заяви про реєстрацiю
або внесення змiн типу II), якщо ранiше вона була
розглянута i затверджена як частина послiдовних
змiн. 2. Змiна не обумовлена непередбаченими обставинами, що виникли в процесi виробництва пакувального матерiалу або зберiгання АФI або дiючої речовини. 3. Будь-якi змiни не повиннi виходити за допустимi межi затвердженої специфiкацiї. 4. Методи випробувань залишилися незмiнними або такi змiни є незначними. 5. Новий метод випробування не належить до нестандартного методу або стандартного методу, що використовується у новий спосiб. |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє. 2. Порiвняльна таблиця вимог затвердженої та запропонованої специфiкацiй. 3. Опис нового методу випробування та данi з його валiдацiї (за необхiдностi). 4. Результати аналiзу для двох серiй первинної упаковки за всiма показниками, зазначеними у специфiкацiї. 5. Обґрунтування вiд власника реєстрацiйного посвiдчення або власника мастер-файла на дiючу речовину, що показник якостi не є суттєвим (за необхiдностi). 6. Обґрунтування вiд власника реєстрацiйного посвiдчення або власника мастер-файла на дiючу речовину нових показникiв якостi та допустимих меж, зазначених у специфiкацiї. |
| 2.1.3.3. Змiна в методах випробування первинної упаковки АФI або дiючої речовини | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) незначнi змiни у затверджених методах випробування | 1, 2, 3 | 1, 2 | IА |
| б) iншi змiни в методах випробування (включаючи замiну або доповнення) | 1, 3, 4 | 1, 2 | IА |
| в) вилучення методу випробування, якщо альтернативний метод випробування вже затверджений | 5 | 1 | IА |
| Умови |
| 1. Вiдповiднi дослiдження з валiдацiї, якi
були проведенi вiдповiдно до чинних фармакопейних
вимог або керiвних принципiв та настанов з
валiдацiї (чинне видання), та результати
пiдтверджують, що затверджена та запропонована
методики - iдентичнi. 2. Методи випробувань залишилися незмiнними (наприклад, змiнилась довжина колонки або температура проведення аналiзу, але тип колонки або методика - незмiннi). 3. Новий метод випробування не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосiб. 4. АФI або дiюча речовина не є речовиною бiологiчного/iмунологiчного походження. 5. Iснує затверджений метод випробування для показника специфiкацiї i цей метод не затверджений при внесеннi змiн типу IА. |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє, якi включають опис методу
випробування та звiт про данi з валiдацiї. 2. Порiвняльнi данi з валiдацiї або, якщо необхiдно, порiвняльнi результати аналiтичних випробувань, якi пiдтверджують iдентичнiсть результатiв, отриманих за затвердженим та новим методами. Це не стосується випадкiв додавання нового методу випробування. |
| 2.1.4. Стабiльнiсть |
| 2.1.4.1. Змiна перiоду повторних випробувань/термiну придатностi або умов зберiгання АФI або дiючої речовини (за наявностi у матерiалах реєстрацiйного досьє сертифiката вiдповiдностi Європейськiй фармакопеї, включає перiод повторного випробування) | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) перiод повторного випробування/термiн придатностi | |||
| зменшення | 1 | 1, 2, 3 | IА |
| збiльшення перiоду повторного випробування на основi екстраполяцiї результатiв дослiдження стабiльностi, проведених не у вiдповiдностi до настанови щодо випробування стабiльностi або керiвних принципiв IСН щодо випробування стабiльностi (крiм АФI або дiючих речовин бiологiчного/iмунологiчного походження) | II | ||
| збiльшення термiну придатностi АФI або дiючої речовини бiологiчного/iмунологiчного походження на основi результатiв дослiджень, виконаних не у вiдповiдностi до затвердженого протоколу стабiльностi | II | ||
| збiльшення або введення перiоду повторного випробування/термiну придатностi на основi результатiв дослiджень у реальному часi | 1, 2, 3 | IБ | |
| б) умови зберiгання | |||
| обмеження умов зберiгання | 1 | 1, 2, 3 | IА |
| змiна умов зберiгання АФI або дiючої речовини бiологiчного/iмунологiчного походження, якщо дослiдження стабiльностi ще не проводились вiдповiдно до затвердженого протоколу стабiльностi | II | ||
| змiна умов зберiгання АФI або дiючої речовини | 1, 2, 3 | IБ |
| Умови |
| 1. Змiна не обумовлена непередбаченими обставинами у процесi виробництва або проблемами щодо стабiльностi. |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє повиннi мiстити результати
дослiджень стабiльностi, представленi у реальному
часi i проведенi вiдповiдно до вимог настанови щодо
випробування стабiльностi або керiвних принципiв
IСН щодо випробування стабiльностi (чинне
видання), принаймнi для двох (трьох - для
лiкарських засобiв бiологiчного походження)
дослiдно-промислових або промислових серiй АФI
або дiючої речовини у затвердженiй упаковцi з
вiдповiдним перiодом повторного випробування у
визначених умовах зберiгання. 2. Пiдтвердження, що дослiдження стабiльностi виконанi вiдповiдно до затвердженого протоколу стабiльностi. Дослiдження повиннi пiдтвердити вiдповiднiсть специфiкацiй. 3. Копiї затверджених специфiкацiй на АФI або дiючу речовину. |
| 2.1.5. Проектний простiр |
| 2.1.5.1. Введення нового проектного простору або розширення затвердженого для АФI або дiючої речовини щодо: | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) одного елементу (блок, частина) виробничого процесу АФI або дiючої речовини, включаючи контроль у процесi виробництва та/або методи випробування | 1, 2, 3 | II | |
| б) методiв випробувань для вихiдних матерiалiв/реагентiв/промiжних продуктiв та/або АФI або дiючої речовини | 1, 2, 3 | II |
| Документацiя |
| 1. Проектний простiр був розроблений
вiдповiдно до європейських та мiжнародних
наукових керiвництв. Результати дослiджень
розробки лiкарського засобу, виробничого процесу
або методiв випробувань (наприклад, необхiдно
вивчити взаємодiю рiзних параметрiв, що формують
проектний простiр, включаючи дослiдження оцiнки
ризику та багатомiрнi дослiдження, якщо необхiдно),
якi демонструють, що функцiональна взаємодiя
властивостей матерiалу та параметрiв процесу, якi
можуть впливати на критичнi характеристики
якостi АФI або дiючої речовини, була досягнута. 2. Опис проектного простору у формi таблицi, включаючи змiннi (властивостi матерiалу та параметри процесу, якщо необхiдно) та їх запропонованi дiапазони. 3. Змiни до вiдповiдних роздiлiв матерiалiв реєстрацiйного досьє. |
| 2.1.5.2. Внесення змiн пiсля затвердження протоколу управлiння для АФI або дiючої речовини | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| 1, 2 | II |
| Документацiя |
| 1. Детальний опис запропонованої змiни. 2. Протокол управлiння змiнами для АФI або дiючої речовини. |
| 2.1.5.3. Вилучення затвердженого протоколу управлiння змiнами для АФI або дiючої речовини | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| 1 | 1 | IА |
| Умови |
| 1. Вилучення, не спричинене непередбаченими обставинами або результатами випробувань, що виходять за межi специфiкацiй пiд час внесення змiни, описаної у протоколi. |
| Документацiя |
| 1. Обґрунтування запропонованого вилучення. |
| 2.2. Готовий лiкарський засiб |
| 2.2.1. Опис та склад |
| 2.2.1.1. Змiна або додавання штампiв, потовщень або iнших маркувань, уключаючи замiну або додавання фарб для маркування лiкарського засобу | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) змiни штампiв, потовщень або iнших маркувань | 1, 2, 3 | 1, 2 | IА |
| б) змiна риски, призначеної для подiлу таблетки на рiвнi дози | 1, 2, 3 | IБ |
| Умови |
| 1. Специфiкацiї при випуску i наприкiнцi
термiну придатностi готового лiкарського засобу
не змiнилися (крiм зовнiшнього вигляду). 2. Будь-яка фарба повинна вiдповiдати вимогам до фармацевтичної продукцiї. 3. Риски не призначено для роздiлу на рiвнi дози. |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє, включаючи детальне
зображення або опис затвердженого та
запропонованого вигляду лiкарського засобу, а
також оновлену коротку характеристику
лiкарського засобу (за необхiдностi). 2. Зразки готового лiкарського засобу (за необхiдностi). 3. Результати фармакопейних випробувань, що пiдтверджують вiдповiднiсть характеристик/ правильного дозування лiкарського засобу. |
| 2.2.1.2. Змiна форми або розмiрiв лiкарської форми | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) таблетки з негайним вивiльненням, капсули, супозиторiї та песарiї | 1, 2, 3, 4 | 1, 4 | IА |
| б) лiкарськi форми, стiйкi до дiї шлункового соку, з модифiкованим або пролонгованим вивiльненням та дiлимi таблетки, призначенi для подiлу на рiвнi дози | 1, 2, 3, 4, 5 | IБ |
| Умови |
| 1. Порiвняння профiлiв розчинення
лiкарського засобу з новими та затвердженими
формою або розмiрами (за необхiдностi). Для
рослинних лiкарських засобiв, коли випробування
на розчинення не може бути проведене, - данi щодо
розпадання. 2. Специфiкацiї при випуску та наприкiнцi термiну придатностi лiкарського засобу не змiнюються (крiм розмiру). 3. Якiсний та кiлькiсний склад, середня маса лiкарського засобу залишаються незмiнними. 4. Змiни не стосуються таблеток з рискою, призначеною для розподiлу на рiвнi дози. |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє, включаючи детальне
зображення затверджених та запропонованих форми
або розмiру, а також оновлену коротку
характеристику лiкарського засобу (за
необхiдностi). 2. Порiвняльнi данi дослiджень для принаймнi однiєї дослiдно-промислової серiї лiкарського засобу, якi пiдтверджують вiдсутнiсть змiн у профiлi розчинення (згiдно з настановами та керiвними принципами щодо дослiдження бiодоступностi) для нових та затверджених розмiру або форми. Для рослинних лiкарських засобiв можуть бути прийнятними данi щодо розпадання. 3. Обґрунтування вiдсутностi необхiдностi проведення нового дослiдження з еквiвалентностi вiдповiдно до вимог настанов та керiвних принципiв щодо дослiдження бiодоступностi. 4. Зразки готового лiкарського засобу з новими розмiром або формою (за потреби). 5. Результати фармакопейних випробувань, що пiдтверджують вiдповiднiсть характеристик/правильного дозування лiкарського засобу. |
| 2.2.1.3. Змiна у складi (допомiжних речовинах) готового лiкарського засобу | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) смаковi добавки або барвники | |||
| додавання, вилучення або замiна | 1, 2, 3, 4, 5, 6, 7, 9 | 1, 2, 4, 5, 6 | IА |
| збiльшення або зменшення | 1, 2, 3, 4 | 1, 2, 4 | IА |
| б) iншi допомiжнi речовини | II | ||
| будь-яка незначна змiна кiлькiсного складу допомiжних речовин у готовому лiкарському засобi | 1, 2, 4, 8, 9, 10 | 1, 2, 7 | IА |
| якiснi або кiлькiснi змiни щодо однiєї або декiлькох допомiжних речовин, якi можуть значно вплинути на безпеку, якiсть або ефективнiсть готового лiкарського засобу | II | ||
| змiна у лiкарському засобi бiологiчного/ iмунологiчного походження | II | ||
| будь-яка нова допомiжна речовина, що включає використання матерiалiв людського або тваринного походження, для яких вимагається оцiнка даних з вiрусної безпеки або ризику передачi збудникiв губчатої енцефалопатiї | II | ||
| змiна, яка пiдтверджується дослiдженнями з еквiвалентностi | II | ||
| замiна однiєї допомiжної речовини на iншу з тими самими функцiональними характеристиками та на тому самому рiвнi | 1, 3, 4, 5, 6, 7, 8, 9 | IБ |
| Умови |
| 1. Не вiдбулося будь-яких змiн
функцiональних характеристик лiкарської форми,
наприклад часу розпадання, профiлю розчинення. 2. Будь-яке незначне коригування складу для збереження загальної маси повинно проводитись щодо допомiжної речовини, що становить основну частину складу готового лiкарського засобу. 3. Специфiкацiї готового лiкарського засобу змiнилися тiльки у частинi зовнiшнього вигляду/запаху/смаку i за потреби вилучено показник якостi щодо iдентифiкацiї. 4. Розпочато дослiдження стабiльностi вiдповiдно до вимог настанови щодо випробування стабiльностi або керiвних принципiв IСН щодо випробування стабiльностi (чинне видання) (iз зазначенням номерiв серiй) для принаймнi двох дослiдно-промислових або промислових серiй iз задовiльними результатами стабiльностi протягом трьох мiсяцiв (на час подання змiн типу IА або IБ), якi демонструють подiбнiсть профiлю стабiльностi до вже затвердженого. Заявник гарантує, що дослiдження будуть завершенi i одразу пiсля завершення дослiджень у разi невiдповiдностi специфiкацiям або за наявної ймовiрностi вiдхилень вiд специфiкацiй наприкiнцi термiну придатностi данi будуть поданi до Центру (з вiдповiдною пропозицiєю). Крiм того, за потреби мають бути проведенi дослiдження фотостабiльностi готового лiкарського засобу. 5. Будь-якi новi компоненти повиннi вiдповiдати встановленим вимогам. 6. Будь-який новий компонент не повинен передбачати використання матерiалiв людського або тваринного походження, для яких необхiдна оцiнка вiрусної безпеки або ризику передачi збудникiв губчатої енцефалопатiї. 7. Змiна не впливає на вiдмiнностi мiж дозуваннями та не спричиняє негативного впливу на смакову прийнятнiсть педiатричних форм (за потреби). 8. Профiль розчинення принаймнi двох дослiдно-промислових серiй запропонованого складу та затвердженого складу лiкарського засобу демонструє вiдсутнiсть будь-яких вiдмiнностей (згiдно з настановами та керiвними принципами щодо дослiдження бiодоступностi). Для рослинних лiкарських засобiв, коли випробування на розчинення не може бути проведене, - данi щодо розпадання. 9. Змiна не обумовлена проблемами зi стабiльнiстю та/або не повинна спричиняти можливi проблеми, пов'язанi з безпекою, тобто розходження мiж дозуваннями. 10. Лiкарський засiб не належить до препаратiв бiологiчного/iмунологiчного походження. |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє (включаючи метод
iдентифiкацiї будь-якого нового барвника за
потреби), а також оновленi: коротка
характеристика лiкарського засобу, iнструкцiя для
медичного застосування та пропозицiї до
маркування на упаковцi (за необхiдностi). 2. Пiдтвердження, що дослiдження стабiльностi розпочато вiдповiдно до вимог настанови щодо випробування стабiльностi або керiвних принципiв IСН щодо випробування стабiльностi (чинне видання) (iз зазначенням номерiв серiй) та пiдтвердження за необхiдностi, що на момент внесення змiн заявником було надано мiнiмум, який вимагається, даних щодо стабiльностi, а також наявнi данi не вказували на будь-яку проблему, пов'язану зi стабiльнiстю. Мають бути наданi гарантiї, що дослiдження будуть завершенi, i одразу пiсля завершення дослiджень у разi невiдповiдностi специфiкацiям або наявної ймовiрностi вiдхилень вiд специфiкацiй наприкiнцi термiну придатностi данi будуть поданi до Центру (iз запропонованими заходами). 3. Результати дослiдження стабiльностi, розпочатi вiдповiдно до вимог настанови щодо випробування стабiльностi або керiвних принципiв IСН щодо випробування стабiльностi (чинне видання), за вiдповiдними параметрами принаймнi для двох дослiдно-промислових або промислових серiй АФI або дiючої речовини iз задовiльними показниками стабiльностi принаймнi за три мiсяцi. При цьому мають бути наданi гарантiї, що дослiдження будуть завершенi, i одразу пiсля завершення дослiджень у разi невiдповiдностi специфiкацiям або наявної ймовiрностi вiдхилень вiд специфiкацiй наприкiнцi термiну придатностi данi будуть поданi до Центру (iз запропонованими заходами). 4. Зразок готового лiкарського засобу з новим складом (за потреби). 5. Сертифiкат вiдповiдностi Європейськiй фармакопеї щодо губчатої енцефалопатiї на будь-який новий вихiдний матерiал тваринного походження або, якщо необхiдно, документальне пiдтвердження того, що вихiдне джерело одержання допомiжної речовини, яке має ризик передачi збудникiв губчатої енцефалопатiї, попередньо оцiнено вповноваженими органами країни-виробника та вiдповiдає керiвництву з мiнiмiзацiї ризику передачi збудникiв губчатої енцефалопатiї тварин з лiкарськими засобами. Iнформацiя повинна мiстити: назву виробника, види та тканини тварин, з яких було одержано вихiдний матерiал, назву країни-постачальника тваринної сировини, її використання. 6. Данi, якi пiдтверджують, що нова допомiжна речовина не впливає на методи контролю готового лiкарського засобу, визначенi у специфiкацiї (за потреби). 7. Обґрунтування змiни/вибору допомiжних речовин тощо, що має бути представлено вiдповiдно до фармацевтичної розробки (включаючи стабiльнiсть та антимiкробнi консерванти, за необхiдностi). 8. Для твердих лiкарських форм порiвняльнi данi щодо профiлю розчинення принаймнi для двох дослiдно-промислових серiй лiкарського засобу з новим та затвердженим складом. Для рослинних лiкарських засобiв можуть бути прийнятними данi щодо розпадання. 9. Обґрунтування вiдсутностi необхiдностi проведення нового дослiдження еквiвалентностi вiдповiдно до вимог настанов або керiвництв з дослiдження бiодоступностi та бiоеквiвалентностi (чинне видання). |
| 2.2.1.4. Змiна маси покриття лiкарських форм для перорального застосування або змiна маси оболонки капсул | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) твердi лiкарськi форми для перорального застосування | 1, 2, 3, 4 | 1, 2 | IА |
| б) лiкарськi форми, стiйкi до дiї шлункового соку, з модифiкованим вивiльненням або пролонгованої дiї, для яких покриття є вирiшальним чинником механiзму вивiльнення | II |
| Умови |
| 1. Вiдсутнiсть змiн у профiлi розчинення
принаймнi двох дослiдно-промислових серiй
лiкарського засобу iз запропонованим та
затвердженим складом. Для рослинних лiкарських
засобiв, коли випробування на розчинення не може
бути проведене, - данi щодо розпадання. 2. Покриття не є вирiшальним чинником механiзму вивiльнення. 3. Специфiкацiї готового лiкарського засобу змiнено тiльки щодо маси i розмiру (за необхiдностi). 4. Дослiдження стабiльностi розпочато вiдповiдно до вимог настанови щодо випробування стабiльностi або керiвних принципiв IСН щодо випробування стабiльностi (чинне видання) для принаймнi двох дослiдно-промислових або промислових серiй iз задовiльними результатами стабiльностi протягом трьох мiсяцiв або бiльше. Заявник гарантує, що дослiдження будуть завершенi, i одразу пiсля завершення дослiджень у разi невiдповiдностi специфiкацiям або за наявної ймовiрностi вiдхилень вiд специфiкацiй наприкiнцi термiну придатностi данi будуть поданi до Центру (з вiдповiдною пропозицiєю). |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє. 2. Пiдтвердження, що дослiдження стабiльностi розпочато вiдповiдно до вимог настанови щодо випробування стабiльностi або керiвних принципiв IСН щодо випробування стабiльностi (чинне видання) (iз зазначенням номерiв серiй), та пiдтвердження за необхiдностi, що на момент внесення змiн заявником було надано мiнiмум, який вимагається, даних щодо стабiльностi. Мають бути наданi гарантiї того, що дослiдження будуть завершенi, i одразу пiсля завершення дослiджень у разi невiдповiдностi специфiкацiям або наявної ймовiрностi вiдхилень вiд специфiкацiй наприкiнцi термiну придатностi данi будуть поданi до Центру (iз запропонованими заходами). Крiм того, у вiдповiдних випадках мають бути проведенi дослiдження фотостабiльностi готового лiкарського засобу. |
| 2.2.1.5. Змiна концентрацiї в окремiй дозi багатодозового лiкарського засобу для парентерального застосування, коли кiлькiсть дiючої речовини на одиницю дози (тобто сила дiї) не змiнюється | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| II |
| 2.2.1.6. Вилучення контейнера з розчинником з упаковки | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| 1, 2 | IБ |
| Документацiя |
| 1. Обґрунтування вилучення, включаючи
заяву щодо альтернативних шляхiв отримання
розчинника, необхiдного для безпечного та
ефективного застосування лiкарського засобу. 2. Оновленi: коротка характеристика лiкарського засобу, iнструкцiя для медичного застосування та пропозицiї до маркування на упаковцi. |
| 2.2.2. Виробництво |
| 2.2.2.1. Замiна або введення додаткової дiльницi виробництва для частини або всього виробничого процесу готового лiкарського засобу | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) дiльниця для вторинного пакування | 1, 2 | 1, 3, 8 | IА |
| б) дiльниця для первинного пакування | 1, 2, 3, 4, 5 | 1, 2, 3, 4, 8, 9 | IА |
| в) дiльниця, на якiй проводяться будь-якi виробничi стадiї, за винятком випуску серiй, проведення контролю якостi та вторинного пакування, для лiкарських засобiв бiологiчного/iмунологiчного походження | II | ||
| г) дiльниця, яка вимагає проведення первинної перевiрки виробництва або перевiрки виробництва конкретного готового лiкарського засобу | II | ||
| ґ) дiльниця, на якiй проводяться будь-якi виробничi стадiї, за винятком випуску серiй, контролю якостi, первинного та вторинного пакування, для нестерильних лiкарських засобiв | 1, 2, 3, 4, 5, 6, 7, 8, 9 | IБ | |
| д) дiльниця, на якiй проводяться будь-якi виробничi стадiї, за винятком випуску серiї, контролю якостi та вторинного пакування для стерильних лiкарських засобiв, що виробленi з використанням асептичного методу, за винятком лiкарських засобiв бiологiчного/iмунологiчного походження | 1, 2, 3, 4, 5, 6, 7, 8 | IБ |
| Умови |
| 1. Задовiльнi результати перевiрки
виробництва за останнi три роки, проведеної
компетентними уповноваженими органами держав,
якi входять до PIC/s, Iнспекцiєю ВООЗ або
уповноваженим органом України. 2. Дiльниця має дозвiл (лiцензiю) на виробництво вiдповiдних лiкарських форм або лiкарського засобу. 3. Цей лiкарський засiб є нестерильним. 4. У разi необхiдностi, наприклад, для суспензiй та емульсiй - проведена схема валiдацiї процесу або успiшно виконана валiдацiя виробництва на новiй дiльницi вiдповiдно до затвердженого протоколу принаймнi на трьох промислових серiях. 5. Цей лiкарський засiб не є лiкарським засобом бiологiчного/iмунологiчного походження. |
| Документацiя |
| 1. Пiдтвердження, що запропонована
дiльниця виробництва має вiдповiдний дозвiл
(лiцензiю) на виробництво лiкарської форми або
лiкарського засобу, а саме: для виробничої дiльницi в Українi - копiя чинної лiцензiї на виробництво; копiя лiцензiї на виробництво (якщо згiдно iз законодавством країни виробника лiцензiя на виробництво iснує лише в електронному виглядi (наприклад, США), має бути надана роздрукiвка iз посиланням на вiдповiдний офiцiйний сайт, засвiдчена пiдписом/печаткою заявника) або iншого дозвiльного документа на виробництво заявленої лiкарської форми у країнi виробника; засвiдчена копiя документа, що пiдтверджує вiдповiднiсть виробництва лiкарського засобу вимогам GMP, виданого Держлiкслужбою вiдповiдно до вимог наказу Мiнiстерства охорони здоров'я України вiд 27 грудня 2012 року N 1130 "Про затвердження Порядку проведення пiдтвердження вiдповiдностi умов виробництва лiкарських засобiв вимогам належної виробничої практики", зареєстрованого у Мiнiстерствi юстицiї України 21 сiчня 2013 року за N 133/22665, або гарантiйний лист заявника щодо надання такого документа протягом строку проведення спецiалiзованої експертизи. 2. Iнформацiя щодо кiлькостi (і 3), номерiв, розмiру та дати виробництва серiй, якi використовуються у дослiдженнях з валiдацiї, данi з валiдацiї або протокол (схема) валiдацiї (за потреби). 3. Заява про внесення змiн має чiтко визначати затвердженого та запропонованого виробникiв готового лiкарського засобу, як зазначено в пунктi 2.5 заяви про державну реєстрацiю лiкарського засобу, форма якої наведена у додатку 1 до Порядку. 4. Копiї затверджених специфiкацiй при випуску та наприкiнцi термiну придатностi (за потреби). 5. Порiвняльнi данi аналiзiв однiєї промислової та двох дослiдно-промислових серiй (або двох промислових серiй), вироблених на запропонованiй дiльницi, та трьох серiй, вироблених на затвердженiй дiльницi. Данi про наступнi двi промисловi серiї мають бути наданi до Центру на вимогу або у разi невiдповiдностi специфiкацiям (з вiдповiдною пропозицiєю). 6. Для м'яких та рiдких лiкарських форм, у яких АФI або дiюча речовина присутня у нерозчиненiй формi, мають бути наданi необхiднi данi з валiдацiї, мiкроскопiчне зображення розподiлу та морфологiя часток. 7. Якщо нова виробнича дiльниця використовує АФI або дiючу речовину як вихiдний матерiал - заява УО, яка вiдповiдає за випуск серiї на дiльницi, що АФI або дiюча речовина виробляється згiдно з вимогами належної виробничої практики щодо вихiдних матерiалiв (чинне видання). 8. Змiни до вiдповiдних роздiлiв матерiалiв реєстрацiйного досьє. 9. Якщо виробництво готового лiкарського засобу та його первинне пакування здiйснюється на рiзних виробничих дiльницях, слiд визначити умови транспортування та зберiгання готового лiкарського засобу у формi in bulk та провести дослiдження з їх валiдацiї. |
| Примiтка. Виробники повиннi використовувати у якостi вихiдних матерiалiв тiльки тi АФI або дiючi речовини, що виробляються вiдповiдно до вимог належної виробничої практики, тому кожний виробник, що використовує АФI або дiючi речовини у якостi вихiдного матерiалу, повинен надати таку заяву. Крiм того, оскiльки УО, вiдповiдальна за випуск серiї, несе вiдповiдальнiсть за серiю, у разi, коли контроль серiї вiдбувається на iншiй дiльницi, необхiдна додаткова заява щодо вiдповiдностi вимогам належної виробничої практики вiд УО, вiдповiдальної за контроль. У випадках, коли виробником виступає одна особа, слiд надавати тiльки одну заяву. Якщо виробникiв бiльше одного, заявник може надати тiльки одну заяву, пiдписану однiєю УО, за умови, що: iз заяви зрозумiло, що вона складена вiд iменi всiх задiяних УО; є у наявностi письмовий контракт мiж заявником та виробником, у якому чiтко визначенi обов'язки кожної зi сторiн, а УО, що надає заяву, зазначена у контрактi як особа, що несе вiдповiдальнiсть за дотримання вимог GMP при виробництвi АФI або дiючої речовини. Заявником пiдтверджується, що вiн користується послугами принаймнi однiєї УО. Не можуть бути прийнятi заяви вiд персоналу, який найнятий виробниками в iнших країнах, включаючи країни з мiжнародними договорами про взаємне визнання результатiв iнспектування на вiдповiднiсть виробництва вимогам належної виробничої практики. Заява не вимагається для кровi або компонентiв кровi, що повиннi вiдповiдати вимогам Директиви 2002/98/ЄС Європейського парламенту та Ради вiд 27 сiчня 2003 року про встановлення стандартiв якостi та безпеки для збору, тестування, обробки, зберiгання та розподiлу людської кровi та її компонентiв, а також поправок до Директиви 2001/83/ЄС. |
| 2.2.2.2. Змiни, що стосуються випуску серiї та контролю якостi готового лiкарського засобу | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) замiна або додавання дiльницi, на якiй здiйснюється контроль/випробування серiї | 2, 3, 4 | 1, 2, 5 | IА |
| б) замiна або додавання виробника, вiдповiдального за випуск серiї | |||
| не включаючи контроль/випробування серiї | 1, 2 | 1, 2, 3, 4, 5 | IА |
| включаючи контроль/випробування серiї | 1, 2, 3, 4 | 1, 2, 3, 4, 5 | IА |
| включаючи контроль/випробування серiї для лiкарського засобу бiологiчного/iмунологiчного походження та один з методiв аналiзу, що застосовується на дiльницi, що є бiологiчним/iмунологiчним/ iмунохiмiчним методом | II |
| Умови |
| 1. Виробник (дiльниця) повинен мати
вiдповiдний дозвiл (лiцензiю). 2. Лiкарський засiб не є лiкарським засобом бiологiчного/iмунологiчного походження. 3. Перенесення iз затвердженої до нової дiльницi або лабораторiї має бути успiшно виконано. |
| Документацiя |
| 1. Пiдтвердження, що запропонована
дiльниця виробництва має вiдповiдний дозвiл
(лiцензiю) на виробництво лiкарської форми або
лiкарського засобу, а саме: для виробничої дiльницi в Українi - копiя чинної лiцензiї на виробництво; копiя лiцензiї на виробництво (якщо згiдно iз законодавством країни виробника лiцензiя на виробництво iснує лише в електронному виглядi (наприклад, США), має бути надана роздрукiвка iз посиланням на вiдповiдний офiцiйний сайт, засвiдчена пiдписом/печаткою заявника) або iншого дозвiльного документа на виробництво заявленої лiкарської форми у країнi виробника; засвiдчена копiя документа, що пiдтверджує вiдповiднiсть виробництва лiкарського засобу вимогам GMP, виданого Держлiкслужбою вiдповiдно до вимог наказу Мiнiстерства охорони здоров'я України вiд 27 грудня 2012 року N 1130 "Про затвердження Порядку проведення пiдтвердження вiдповiдностi умов виробництва лiкарських засобiв вимогам належної виробничої практики", зареєстрованого у Мiнiстерствi юстицiї України 21 сiчня 2013 року за N 133/22665, або гарантiйного листа заявника щодо надання такого документа протягом строку проведення спецiалiзованої експертизи. 2. Заява про внесення змiн має чiтко визначати затвердженого та запропонованого виробникiв готового лiкарського засобу, як зазначено в пунктi 2.5 заяви про державну реєстрацiю лiкарського засобу, форма якої наведена у додатку 1 до Порядку. 3. Заява Уповноваженої особи, яка вiдповiдає за контроль якостi серiї, що АФI або дiюча речовина виробляється згiдно з вимогами належної виробничої практики щодо вихiдних матерiалiв (чинне видання). Узагальнена заява може бути надана за умов, викладених у пiдпунктi 2.2.2.1 цього додатка. 4. Змiни до вiдповiдних роздiлiв матерiалiв реєстрацiйного досьє, включаючи оновлену коротку характеристику лiкарського засобу, iнструкцiю для медичного застосування та пропозицiї до маркування на упаковцi (за необхiдностi). |
| 2.2.2.3. Змiни у процесi виробництва готового лiкарського засобу | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) незначна змiна у процесi виробництва твердої лiкарської форми для перорального застосування з негайним вивiльненням або розчинiв для перорального застосування | 1, 2, 3, 4, 5, 6, 7 | 1, 3, 4, 6, 7, 8 | IА |
| б) змiна у процесi виробництва, яка може мати iстотний влив на якiсть, безпеку та ефективнiсть лiкарського засобу | II | ||
| в) лiкарський засiб є лiкарським засобом бiологiчного/iмунологiчного походження та змiна вимагає проведення порiвняльних дослiджень | II | ||
| г) ведення нестандартного методу кiнцевої стерилiзацiї | II | ||
| ґ) введення або збiльшення припустимого надлишку дiючої речовини | II | ||
| д) незначна змiна у процесi виробництва водної суспензiї для перорального застосування | 1, 2, 4, 6, 7, 8 | IБ |
| Умови |
| 1. Немає жодних змiн у якiсних або
кiлькiсних показниках профiлю домiшок або у
фiзико-хiмiчних властивостях. 2. Цей лiкарський засiб не є лiкарським засобом бiологiчного/iмунологiчного або рослинного походження. 3. Виробничий процес, який включає окремi стадiї, залишається таким самим, наприклад, обробка промiжних продуктiв, i немає жодних змiн у будь-якому розчиннику, що використовується у процесi виробництва. 4. Затверджений виробничий процес має контролюватися вiдповiдними методами i цi методи не потребують жодних змiн (розширення або вилучення допустимих меж). 5. Специфiкацiї на готовий лiкарський засiб або промiжнi продукти залишаються незмiнними. 6. Новий виробничий процес повинен забезпечити виготовлення аналогiчного попередньому лiкарського засобу за усiма показниками якостi, безпеки та ефективностi. 7. Розпочато дослiдження стабiльностi вiдповiдно до вимог настанови щодо випробування стабiльностi або керiвних принципiв IСН щодо випробування стабiльностi (чинне видання) для принаймнi однiєї дослiдно-промислової або промислової серiї iз задовiльними результатами стабiльностi протягом трьох мiсяцiв або бiльше. Заявник гарантує, що дослiдження будуть завершенi, i одразу пiсля завершення дослiджень у разi невiдповiдностi специфiкацiям або за наявної ймовiрностi вiдхилень вiд специфiкацiй наприкiнцi термiну придатностi данi будуть поданi до Центру (з вiдповiдною пропозицiєю). |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє, включаючи порiвняння
затвердженого та запропонованого виробничого
процесу. 2. Для м'яких та рiдких лiкарських форм, у яких дiюча речовина мiститься у нерозчиннiй формi, вiдповiднi данi з валiдацiї змiн, включаючи мiкроскопiю часток для перевiрки видимих змiн у морфологiї; порiвняльнi данi гранулометричного складу за вiдповiдним методом. 3. Для твердих лiкарських форм - данi, що пiдтверджують вiдсутнiсть змiн у профiлi розчинення однiєї промислової серiї, виробленої за змiненою технологiєю порiвняно з останнiми трьома серiями, виробленими за узгодженою технологiєю. Данi про наступнi двi повнi промисловi серiї мають бути наданi на вимогу або у разi невiдповiдностi специфiкацiям буде повiдомлено Центр (з вiдповiдною пропозицiєю). Для рослинних лiкарських засобiв можуть бути прийнятними данi щодо розпадання. 4. Обґрунтування вiдсутностi необхiдностi проведення дослiджень еквiвалентностi згiдно з настановами або керiвництвами з дослiдження бiодоступностi (чинне видання). 5. У разi змiни у процесi стерилiзацiї необхiдно надати данi з валiдацiї. 6. Копiя затверджених специфiкацiй при випуску та наприкiнцi термiну придатностi. 7. Данi аналiзiв (у виглядi порiвняльної таблицi) принаймнi однiєї промислової серiї, виробленої за узгодженою та запропонованою технологiями. Данi про наступнi двi повнi промисловi серiї мають бути наданi на вимогу або у разi невiдповiдностi специфiкацiям буде повiдомлено Центр (з вiдповiдною пропозицiєю). 8. Пiдтвердження, що дослiдження стабiльностi розпочато вiдповiдно до вимог настанови щодо випробування стабiльностi або керiвних принципiв IСН щодо випробування стабiльностi (чинне видання) (iз зазначенням номерiв серiй) для принаймнi однiєї дослiдно-промислової або промислової серiї iз задовiльними результатами стабiльностi протягом трьох мiсяцiв або бiльше та профiлем стабiльностi, подiбним до профiлю стабiльностi лiкарського засобу, виробленого за узгодженою технологiєю. Мають бути наданi гарантiї того, що дослiдження будуть завершенi, i одразу пiсля завершення дослiджень у разi невiдповiдностi специфiкацiям або наявної ймовiрностi вiдхилень вiд специфiкацiй наприкiнцi термiну придатностi данi будуть поданi до Центру (iз запропонованими заходами). |
| 2.2.2.4. Змiна розмiру серiї (включаючи дiапазон розмiру серiї) готового лiкарського засобу | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) збiльшення до 10 разiв порiвняно iз затвердженим розмiром серiї | 1, 2, 3, 4, 5, 7 | 1, 4 | IА |
| б) зменшення до 10 разiв порiвняно iз затвердженим розмiром серiї | 1, 2, 3, 4, 5, 6 | 1, 4 | IА |
| в) змiна вимагає оцiнки порiвнянностi (проведення порiвняльних дослiджень) лiкарського засобу бiологiчного/iмунологiчного походження | II | ||
| г) змiна стосується всiх iнших лiкарських форм сукупного (комплексного) виробничого процесу | II | ||
| ґ) збiльшення розмiру серiї бiльш нiж у 10 разiв порiвняно iз затвердженим розмiром для твердих лiкарських форм з негайним вивiльненням | 1, 2, 3, 4, 5, 6 | IБ | |
| д) масштаб для лiкарського засобу бiологiчного/iмунологiчного походження збiльшився/зменшився без змiни виробничого процесу (наприклад, дублювання лiнiї) | 1, 2, 3, 4, 5, 6 | IБ |
| Умови |
| 1. Змiна не впливає на вiдтворюванiсть
та/або постiйнiсть лiкарського засобу. 2. Змiна стосується тiльки твердих лiкарських форм з негайним вивiльненням для перорального застосування або нестерильних рiдких лiкарських форм. 3. Будь-якi змiни методу виробництва та/або контролю у процесi виробництва спричиненi змiною розмiру серiї, наприклад, використанням обладнання iншої продуктивностi. 4. Iснує схема валiдацiї або валiдацiя виробництва була успiшно проведена згiдно iз затвердженим протоколом з використанням принаймнi трьох серiй лiкарського засобу нового розмiру вiдповiдно до настанов або керiвництв з валiдацiї виробництва (чинне видання). 5. Цей лiкарський засiб не є лiкарським засобом бiологiчного/iмунологiчного походження. 6. Змiна не обумовлена непередбаченими обставинами у процесi виробництва або пересторогами щодо стабiльностi. 7. Попереднiй розмiр серiї не був затверджений пiд час внесення змiни типу IА. |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє. 2. Данi аналiзiв (у виглядi порiвняльної таблицi) принаймнi однiєї промислової серiї затвердженого та запропонованого розмiрiв. Данi про наступнi двi повнi промисловi серiї мають бути наданi на вимогу або у разi невiдповiдностi специфiкацiям буде повiдомлено Центр (з вiдповiдною пропозицiєю). 3. Копiї затверджених специфiкацiй при випуску та наприкiнцi термiну придатностi. 4. Iнформацiя щодо кiлькостi (і 3), номерiв, розмiрiв серiй лiкарських засобiв та дат їх виробництва, що використовуються у дослiдженнях з валiдацiї, або схема валiдацiї. 5. Данi з валiдацiї. 6. Пiдтвердження, що дослiдження стабiльностi розпочатi вiдповiдно до вимог настанови щодо випробування стабiльностi або керiвних принципiв IСН щодо випробування стабiльностi (чинне видання) (iз зазначенням кiлькостi та номерiв серiй) для принаймнi однiєї дослiдно-промислової або промислової серiї iз задовiльними результатами стабiльностi протягом трьох мiсяцiв або бiльше. Мають бути наданi гарантiї того, що дослiдження будуть завершенi, i одразу пiсля завершення дослiджень у разi невiдповiдностi специфiкацiям або наявної ймовiрностi вiдхилень вiд специфiкацiй наприкiнцi термiну придатностi данi будуть поданi до Центру (iз запропонованими заходами). Для лiкарських засобiв бiологiчного/iмунологiчного походження - пiдтвердження вiдсутностi необхiдностi проведення порiвняльних дослiджень. |
| 2.2.2.5. Змiни випробувань або допустимих меж, встановлених у специфiкацiях, пiд час виробництва готового лiкарського засобу | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) звуження допустимих меж | 1, 2, 3, 4 | 1, 2 | IА |
| б) доповнення нового методу випробування та допустимих меж | 1, 2, 5, 6 | 1, 2, 3, 4, 5, 7 | IА |
| в) вилучення несуттєвого випробування | 1, 2 | 1, 2, 6 | IА |
| г) вилучення випробування у процесi виробництва, яке може мати iстотний вплив на загальну якiсть готового лiкарського засобу | II | ||
| ґ) розширення затверджених допустимих меж для показникiв, якi можуть мати iстотний вплив на загальну якiсть готового лiкарського засобу | II | ||
| д) доповнення або замiна випробування за результатами дослiджень з безпеки або якостi | 1, 2, 3, 4, 5, 7 | IБ |
| Умови |
| 1. Змiна не обумовлена попередньою
експертизою, що здiйснювалась для перегляду
допустимих меж специфiкацiй (наприклад,
проведеною пiд час подання заяви про реєстрацiю
або внесення змiн типу II). 2. Змiна не обумовлена непередбаченими обставинами у процесi виробництва, наприклад, утворенням нової неквалiфiкованої домiшки, змiною допустимих меж загального вмiсту домiшок. 3. Будь-яка змiна не повинна виходити за допустимi межi затвердженої специфiкацiї. 4. Метод випробування залишається незмiнним або такi змiни є незначними. 5. Будь-який новий метод випробування не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосiб. 6. Новий метод випробування не належить до бiологiчного/iмунологiчного/iмунохiмiчного методу або методу, у якому використовується бiологiчний реактив для субстанцiї бiологiчного походження (за винятком стандартних фармакопейних мiкробiологiчних методiв). |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє. 2. Порiвняльна таблиця щодо затвердженого та запропонованого випробування та допустимих меж. 3. Опис будь-якого нового методу випробування та данi з його валiдацiї (за необхiдностi). 4. Данi аналiзiв для двох промислових серiй (трьох промислових серiй для лiкарських засобiв бiологiчного походження, якщо не обґрунтовано iнше) готового лiкарського засобу за всiма показниками специфiкацiй. 5. Порiвняльнi данi щодо профiлю розчинення для готового лiкарського засобу принаймнi однiєї дослiдної серiї, виготовленої за затверджених та запропонованих умов (за необхiдностi). Для рослинних лiкарських засобiв можуть бути прийнятнi данi щодо розпадання. 6. Обґрунтування/оцiнка ризику, що пiдтверджує незначнiсть змiненого параметра. 7. Обґрунтування введення нового випробування у процесi виробництва та допустимих меж. |
| 2.2.3. Контроль допомiжних речовин |
| 2.2.3.1. Змiна параметрiв специфiкацiй та/або допустимих меж для допомiжної речовини | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) звуження допустимих меж | 1, 2, 3, 4 | 1, 2 | IА |
| б) доповнення специфiкацiї новим показником з вiдповiдним методом випробування | 1, 2, 5, 6, 7 | 1, 2, 3, 4, 6, 8 | IА |
| в) вилучення iз специфiкацiї незначного показника (наприклад, вилучення застарiлого показника) | 1, 2 | 1, 2, 7 | IА |
| г) змiна знаходиться поза затвердженими допустимими межами специфiкацiй | II | ||
| ґ) вилучення iз специфiкацiї показника, який може мати iстотний вплив на загальну якiсть готового лiкарського засобу | II | ||
| д) доповнення або замiна показника специфiкацiї за результатами дослiджень з безпеки або якостi (за винятком лiкарських засобiв бiологiчного та iмунологiчного походження) | 1, 2, 3, 4, 5, 6, 8 | IБ |
| Умови |
| 1. Змiна не обумовлена попередньою
експертизою, що здiйснювалась для перегляду
допустимих меж специфiкацiй (наприклад,
проведеною пiд час подання заяви про реєстрацiю
або внесення змiн типу II). 2. Змiна не обумовлена непередбаченими обставинами у процесi виробництва, наприклад, утворення нової неквалiфiкованої домiшки; змiна допустимих меж загального вмiсту домiшок. 3. Будь-яка змiна не повинна виходити за допустимi межi затвердженої специфiкацiї. 4. Метод випробування залишається незмiнним або такi змiни є незначними. 5. Будь-який новий метод випробування не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосiб. 6. Новий метод випробування не належить до бiологiчного/iмунологiчного/iмунохiмiчного методу або методу, у якому використовується бiологiчний реактив для субстанцiї бiологiчного походження (за винятком стандартних фармакопейних мiкробiологiчних методiв). 7. Змiни не стосуються домiшки, яка має генотоксичну дiю. |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє. 2. Порiвняльна таблиця щодо затвердженої та запропонованої специфiкацiй. 3. Опис нового методу випробування та данi з його валiдацiї (за необхiдностi). 4. Данi аналiзiв для двох промислових серiй (трьох промислових серiй для лiкарських засобiв бiологiчного походження, якщо не обґрунтовано iнше) готового лiкарського засобу за всiма показниками специфiкацiй. 5. Порiвняльнi данi профiлю розчинення для готового лiкарського засобу принаймнi однiєї дослiдної серiї з допомiжними речовинами, якi вiдповiдають затвердженiй та запропонованiй специфiкацiям (за необхiдностi). Для рослинних лiкарських засобiв можуть бути прийнятнi данi щодо розпадання. 6. Обґрунтування вiдсутностi нових даних з еквiвалентностi згiдно з вимогами настанов та керiвними принципами щодо дослiдження бiодоступностi (чинне видання) (за необхiдностi). 7. Обґрунтування/оцiнка ризику, що пiдтверджує незначнiсть змiненого параметра. 8. Обґрунтування введення нового показника специфiкацiї та допустимих меж. |
| 2.2.3.2. Змiна у методах випробування допомiжної речовини | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) незначнi змiни у затверджених методах випробувань | 1, 2, 3, 4 | 1, 2 | IА |
| б) вилучення методу випробування, якщо альтернативний метод випробування вже затверджений | 5 | 1 | IА |
| в) замiна бiологiчного/iмунологiчного/ iмунохiмiчного методу випробування або методу, у якому використовується бiологiчний реагент | II | ||
| г) iншi змiни у методах випробування (включаючи замiну або додавання) | 1, 2 | IБ |
| Умови |
| 1. Дослiдження з валiдацiї були проведенi
вiдповiдно до чинних фармакопейних вимог або
настанов та керiвних принципiв з валiдацiї (чинне
видання) та результати проведених дослiджень
пiдтверджують, що нова методика - iдентична
затвердженiй. 2. Не було жодних змiн меж загального вмiсту домiшок, не виявлено нових неквалiфiкованих домiшок. 3. Методи випробувань залишилися незмiнними (наприклад, змiнилась довжина колонки або температура проведення аналiзу, але тип колонки або методика - незмiннi). 4. Новий метод випробувань не належить до бiологiчних/iмунологiчних/iмунохiмiчних методiв або методу, при якому використовується бiологiчний реагент для аналiзу субстанцiї бiологiчного походження (за винятком стандартних фармакопейних мiкробiологiчних методiв). 5. Iснує затверджений метод випробування для показника специфiкацiї i цей метод не затверджений при внесеннi змiни типу IА. |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє, включаючи опис методу
аналiзу, звiт про данi з валiдацiї, оновленi
специфiкацiї щодо домiшок (за необхiдностi). 2. Порiвняльнi данi з валiдацiї або, якщо обґрунтовано, порiвняльнi результати аналiзу, якi пiдтверджують, що затверджений та запропонований методи випробування є iдентичними. Ця вимога не стосується випадку додавання нового методу випробування. |
| 2.2.3.3. Замiна джерела одержання допомiжної речовини або реактиву, що становить ризик передачi збудникiв губчатої енцефалопатiї | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) матерiалу, що становить ризик передачi збудникiв губчатої енцефалопатiї, на матерiал рослинного або синтетичного походження | |||
| для допомiжних речовин або реактивiв, якi не використовуються у виробництвi активної субстанцiї бiологiчного/iмунологiчного походження або лiкарського засобу бiологiчного/iмунологiчного походження | 1 | 1 | IА |
| для допомiжних речовин або реактивiв, якi використовуються у виробництвi активної субстанцiї бiологiчного/iмунологiчного походження або лiкарського засобу бiологiчного/iмунологiчного походження | 1, 2 | IБ | |
| б) замiна або додавання речовини, що становить ризик передачi збудникiв губчатої енцефалопатiї, або замiна речовини, що становить ризик передачi збудникiв губчатої енцефалопатiї, на iншу речовину, що становить ризик передачi збудникiв губчатої енцефалопатiї, для якої немає сертифiката вiдповiдностi Європейськiй фармакопеї щодо губчатої енцефалопатiї | II |
| Умови |
| 1. Специфiкацiї при випуску та наприкiнцi термiну придатностi на допомiжну речовину та готовий лiкарський засiб залишаються незмiнними. |
| Документацiя |
| 1. Заява вiд виробника або заявника, яка
пiдтверджує, що допомiжна речовина є речовиною
виключно рослинного або синтетичного
походження. 2. Результати дослiдження еквiвалентностi матерiалiв, їх впливу на виробництво кiнцевої речовини та впливу на характеристики (наприклад, параметри розчинення) готового лiкарського засобу. |
| 2.2.3.4. Змiни в методi синтезу або регенерацiї нефармакопейної допомiжної речовини (за наявностi в досьє) | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) незначнi змiни у методi синтезу або регенерацiї | 1, 2 | 1, 2, 3, 4 | IА |
| б) змiни у специфiкацiї або змiна фiзико-хiмiчних властивостей допомiжної речовини, що може мати вплив на якiсть готового лiкарського засобу | II | ||
| в) допомiжна речовина є речовиною бiологiчного/iмунологiчного походження | II |
| Умови |
| 1. Метод синтезу та специфiкацiї
залишаються незмiнними, вiдсутнi змiни у якiсних i
кiлькiсних показниках профiлю домiшок (за винятком
залишкових розчинникiв, за умови, що вони
вiдповiдають керiвним принципам IСН) або
фiзико-хiмiчних властивостях. 2. За винятком ад'ювантiв для вакцин. |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє. 2. Данi аналiзiв (у формi порiвняльної таблицi) принаймнi двох серiй (мiнiмум дослiдно-промислових) з використанням допомiжної речовини, виготовленої за узгодженим та новим методами виробництва. 3. Порiвняльнi данi щодо профiлю розчинення для готового лiкарського засобу принаймнi двох серiй (мiнiмум дослiдно-промислових) з використанням допомiжної речовини, виготовленої за узгодженим та новим методами виробництва (за необхiдностi). Для рослинних лiкарських засобiв можуть бути прийнятними данi щодо розпадання. 4. Копiї затверджених та нових (за необхiдностi) специфiкацiй на допомiжну речовину. |
| 2.2.4. Контроль готового лiкарського засобу |
| 2.2.4.1. Змiна параметрiв специфiкацiй та/або допустимих меж готового лiкарського засобу | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) звуження допустимих меж | 1, 2, 3, 4 | 1, 2 | IА |
| б) звуження допустимих меж для лiкарського засобу, що пiдлягає офiцiйному випуску серiї | 1, 2, 3, 4 | 1, 2 | IА |
| в) доповнення специфiкацiї новим показником з вiдповiдним методом випробування | 1, 2, 5, 6, 7 | 1, 2, 3, 4, 5, 7 | IА |
| г) вилучення незначного показника (наприклад, вилучення застарiлого показника) | 1, 2 | 1, 2, 6 | IА |
| ґ) змiна знаходиться поза затвердженими допустимими межами | II | ||
| д) вилучення показника, який може мати iстотний вплив на якiсть готового лiкарського засобу | II | ||
| е) доповнення або замiна показника специфiкацiї за результатами дослiджень з безпеки або якостi (за винятком лiкарських засобiв бiологiчного/ iмунологiчного походження) | 1, 2, 3, 4, 5, 7 | IБ |
| Умови |
| 1. Змiна не обумовлена попередньою
експертизою, що здiйснювалась для перегляду
допустимих меж специфiкацiй (наприклад,
проведеною пiд час подання заяви про реєстрацiю
або внесення змiн типу II). 2. Змiна не обумовлена непередбаченими обставинами у процесi виробництва, наприклад, утворенням нової неквалiфiкованої домiшки; змiною допустимих меж загального вмiсту домiшок. 3. Будь-яка змiна не повинна виходити за допустимi межi затвердженої специфiкацiї. 4. Метод випробування залишається незмiнним або такi змiни є незначними. 5. Будь-який новий метод випробування не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосiб. 6. Новий метод випробування не належить до бiологiчного/iмунологiчного/iмунохiмiчного методу або методу, у якому використовується бiологiчний реактив для АФI або дiючої речовини бiологiчного походження (за винятком стандартних фармакопейних мiкробiологiчних методiв). 7. Змiни не стосуються домiшки, яка має генотоксичну дiю. |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє. 2. Порiвняльна таблиця щодо затверджених та запропонованих специфiкацiй. 3. Опис нового методу випробування та данi з його валiдацiї (за необхiдностi). 4. Данi аналiзiв для двох промислових серiй (трьох промислових серiй для лiкарських засобiв бiологiчного походження, якщо не обґрунтовано iнше) готового лiкарського засобу за всiма показниками специфiкацiй. 5. Порiвняльнi данi профiлю розчинення для готового лiкарського засобу принаймнi однiєї дослiдної серiї, якi вiдповiдають затвердженiй та запропонованiй специфiкацiям (за необхiдностi). Для рослинних лiкарських засобiв можуть бути прийнятнi данi щодо розпадання. 6. Обґрунтування/оцiнка ризику, що пiдтверджує незначнiсть змiненого параметра. 7. Обґрунтування введення нового показника та допустимих меж. |
| 2.2.4.2. Змiна у методах випробування готового лiкарського засобу | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) незначна змiна у затверджених методах випробування | 1, 2, 3, 4 | 1, 2 | IА |
| б) вилучення методу випробування, якщо вже затверджено альтернативний метод | 4 | 1 | IА |
| в) змiна у бiологiчному/iмунологiчному/ iмунохiмiчному методi випробування або методi, у якому використовується бiологiчний реагент | II | ||
| г) iншi змiни у методах випробувань (включаючи замiну або доповнення) | 1, 2 | IБ |
| Умови |
| 1. Дослiдження з валiдацiї були проведенi
вiдповiдно до чинних фармакопейних вимог або
настанов, або керiвних принципiв з валiдацiї (чинне
видання) та результати проведених дослiджень
пiдтверджують, що нова методика - iдентична
затвердженiй. 2. Не було жодних змiн меж загального вмiсту домiшок, не виявлено нових неквалiфiкованих домiшок. 3. Методи випробувань залишилися незмiнними (наприклад, змiнилась довжина колонки або температура проведення аналiзу, але тип колонки або методика - незмiннi). 4. Новий метод випробувань не належить до бiологiчних/iмунологiчних/iмунохiмiчних методiв або методу, при якому використовується бiологiчний реагент для аналiзу АФI або дiючої речовини бiологiчного походження (за винятком стандартних фармакопейних мiкробiологiчних методiв). |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє, включаючи опис методу
аналiзу, звiт про данi з валiдацiї, оновленi
специфiкацiї щодо домiшок (якщо необхiдно). 2. Порiвняльнi данi валiдацiї або, якщо обґрунтовано, порiвняльнi результати аналiзу, якi пiдтверджують, що затверджений та запропонований методи випробування є iдентичними. Ця вимога не стосується випадку додавання нового методу випробування. |
| 2.2.4.3. Змiни, якi стосуються виробничого процесу у реальному часi або випуску за параметрами для готового лiкарського засобу | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| II |
| 2.2.5. Система упаковка/укупорка |
| 2.2.5.1. Змiна у первиннiй упаковцi готового лiкарського засобу | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiн |
| а) якiсний та кiлькiсний склад | |||
| твердi лiкарськi форми | 1, 2, 3 | 1, 2, 3, 4, 6 | IА |
| м'якi та нестерильнi рiдкi лiкарськi форми | 1, 2, 3, 5, 6 | IБ | |
| стерильнi лiкарськi засоби та лiкарськi засоби бiологiчного/iмунологiчного походження | II | ||
| змiна стосується зниження ступеня захисту оновленої упаковки, якщо наявнi вiдповiднi змiни в умовах зберiгання та/або скорочення термiну придатностi | II | ||
| б) тип контейнера | |||
| твердi, м'якi та нестерильнi рiдкi лiкарськi форми | 1, 2, 3, 5, 6, 7 | IБ | |
| стерильнi лiкарськi засоби та лiкарськi засоби бiологiчного/iмунологiчного походження | II |
| Умови |
| 1. Змiна стосується лише одного типу
упаковки/контейнера (наприклад, замiна блiстера
на блiстер). 2. Запропонований пакувальний матерiал повинен бути iдентичним затвердженому за вiдповiдними властивостями. 3. Розпочато дослiдження стабiльностi вiдповiдно до вимог настанови щодо випробування стабiльностi або керiвних принципiв IСН щодо випробування стабiльностi (чинне видання) для принаймнi двох дослiдно-промислових або промислових серiй iз задовiльними результатами стабiльностi протягом трьох мiсяцiв або бiльше. Однак, якщо запропонований пакувальний матерiал - бiльш стiйкий, нiж затверджений (наприклад, матерiал блiстера має бiльшу товщину), данi щодо стабiльностi за три мiсяцi ще є недоступними. Мають бути наданi гарантiї того, що дослiдження будуть завершенi, i одразу пiсля завершення дослiджень у разi невiдповiдностi специфiкацiям або наявної ймовiрностi вiдхилень вiд специфiкацiй наприкiнцi термiну придатностi данi будуть поданi до Центру (iз запропонованими заходами). |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє, включаючи оновленi: коротка
характеристика лiкарського засобу, iнструкцiя для
медичного застосування та пропозицiї до
маркування на упаковцi (за необхiдностi). 2. Вiдповiднi данi щодо нової упаковки (порiвняльнi данi про проникнiсть, наприклад для O2, CO2, вологи). 3. Пiдтвердження вiдсутностi (за необхiдностi) будь-якої взаємодiї мiж лiкарським засобом та пакувальним матерiалом (наприклад, не було перенесення компонентiв запропонованого пакувального матерiалу до лiкарського засобу та навпаки), включаючи пiдтвердження того, що пакувальний матерiал вiдповiдає чинним фармакопейним вимогам щодо пакувальних матерiалiв або законодавству ЄС щодо безпеки взаємодiї пластикових пакувальних матерiалiв та упаковок з харчовими продуктами. 4. Заява, що дослiдження стабiльностi розпочато вiдповiдно до вимог настанови щодо випробування стабiльностi або керiвних принципiв IСН щодо випробування стабiльностi (чинне видання) (iз зазначенням номерiв серiй) та (за необхiдностi) що на момент внесення змiн заявником було надано мiнiмум, який вимагається, даних щодо стабiльностi, а також наявнi данi не вказували на будь-яку проблему, пов'язану зi стабiльнiстю. Мають бути наданi гарантiї, що дослiдження будуть завершенi, i у разi невiдповiдностi специфiкацiям або наявної ймовiрностi вiдхилень вiд специфiкацiй наприкiнцi термiну придатностi, одразу пiсля завершення дослiджень данi будуть поданi до Центру (iз запропонованими заходами). 5. Результати дослiдження стабiльностi, якi було проведено вiдповiдно до вимог настанови щодо випробування стабiльностi або керiвних принципiв IСН щодо випробування стабiльностi (чинне видання), за вiдповiдними параметрами принаймнi для двох дослiдно-промислових або промислових серiй лiкарського засобу iз задовiльними показниками щодо стабiльностi принаймнi за три мiсяцi. При цьому мають бути наданi гарантiї того, що дослiдження будуть завершенi, i одразу пiсля завершення дослiджень у разi невiдповiдностi специфiкацiям або наявної ймовiрностi вiдхилень вiд специфiкацiй наприкiнцi термiну придатностi данi будуть поданi до Центру (iз запропонованими заходами). 6. Порiвняльна таблиця вимог затвердженої та запропонованої специфiкацiй (за необхiдностi). 7. Зразки нової системи упаковка/укупорка (за необхiдностi). |
| Примiтка. Будь-яка змiна, що призводить до нової лiкарської форми, потребує подання заяви про державну реєстрацiю лiкарського засобу, форма якої наведена у додатку 1 до Порядку. |
| 2.2.5.2. Змiна параметрiв специфiкацiй та/або допустимих меж первинної упаковки готового лiкарського засобу | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) звуження допустимих меж, визначених у специфiкацiї | 1, 2, 3, 4 | 1, 2 | IА |
| б) доповнення специфiкацiї новим показником з вiдповiдним методом випробування | 1, 2, 5 | 1, 2, 3, 4, 6 | IА |
| в) вилучення незначного показника (наприклад, вилучення застарiлого показника) | 1, 2 | 1, 2, 5 | IА |
| г) додавання або замiна показника за результатами дослiджень з безпеки або якостi | 1, 2, 3, 4, 6 | IБ |
| Умови |
| 1. Змiна не обумовлена попередньою
експертизою, що здiйснювалась для перегляду
параметрiв специфiкацiї (наприклад, проведеною пiд
час подання заяви про реєстрацiю або внесення
змiн типу II). 2. Змiна, не обумовлена непередбаченими обставинами у процесi виробництва. 3. Будь-яка змiна не повинна виходити за допустимi межi затвердженої специфiкацiї. 4. Методи випробування залишаються незмiнними або такi змiни є незначними. 5. Будь-який новий метод випробування не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосiб. |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє. 2. Порiвняльна таблиця вимог затвердженої та запропонованої специфiкацiй. 3. Опис нового методу випробування та данi з його валiдацiї (за необхiдностi). 4. Результати аналiзу для двох серiй первинної упаковки за усiма показниками, зазначеними у специфiкацiї. 5. Обґрунтування того, що показник є незначним. 6. Обґрунтування нового показника якостi та допустимих меж. |
| 2.2.5.3. Змiна у методах випробування первинної упаковки готового лiкарського засобу | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) незначнi змiни у затверджених методах випробувань | 1, 2, 3 | 1, 2 | IА |
| б) iншi змiни у методах випробувань (включаючи замiну або додавання) | 1, 3, 4 | 1, 2 | IА |
| в) вилучення методу випробування, якщо вже затверджений альтернативний | 5 | 1 | IА |
| Умови |
| 1. Дослiдження з валiдацiї були проведенi
вiдповiдно до чинних фармакопейних вимог або
настанов та керiвних принципiв з валiдацiї (чинне
видання) та результати проведених дослiджень
пiдтверджують, що нова методика - iдентична
затвердженiй. 2. Методи випробувань залишилися незмiнними (наприклад, змiнилась довжина колонки або температура проведення аналiзу, але тип колонки або методика - незмiннi). 3. Будь-який новий метод випробувань не належить до нового нестандартного методу або стандартного методу, що використовується у новий спосiб. 4. Субстанцiя/готовий лiкарський засiб не є речовиною/лiкарським засобом бiологiчного/iмунологiчного походження. 5. Iснує затверджений метод випробування для показника специфiкацiї i цей метод не затверджений при внесеннi змiни типу IА. |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє, включаючи опис методу
випробування та звiт з його валiдацiї. 2. Порiвняльнi данi валiдацiї або, якщо обумовлено, порiвняльнi результати аналiзу, якi пiдтверджують iдентичнiсть результатiв, отриманих за затвердженим та новим методами випробувань. Це не стосується випадкiв додавання нового методу випробування. |
| 2.2.5.4. Змiна форми або розмiру контейнера чи закупорювального засобу (первинної упаковки) | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) нестерильнi лiкарськi засоби | 1, 2, 3 | 1, 2, 4 | IА |
| б) змiна форми або розмiру основної частини пакувального матерiалу (вторинної упаковки), що може мати значний вплив на доставку, застосування, безпеку та стабiльнiсть готового лiкарського засобу | II | ||
| в) стерильнi лiкарськi засоби | 1, 2, 3, 4 | IБ |
| Умови |
| 1. Вiдсутнi якiснi або кiлькiснi змiни
складу пакувального матерiалу. 2. Змiна не стосується основної частини пакувального матерiалу, яка впливає на доставку, застосування, безпеку та стабiльнiсть готового лiкарського засобу. 3. У разi змiни вiльного простору над лiкарським засобом або змiни у спiввiдношеннi поверхня/об'єм дослiдження стабiльностi розпочато вiдповiдно до вимог настанови щодо випробування стабiльностi або керiвних принципiв IСН щодо випробування стабiльностi (чинне видання) принаймнi для двох дослiдно-промислових (трьох для лiкарських засобiв бiологiчного/iмунологiчного походження) або промислових серiй лiкарського засобу iз задовiльними показниками щодо стабiльностi принаймнi за три мiсяцi (шiсть мiсяцiв для лiкарських засобiв бiологiчного/iмунологiчного походження). При цьому мають бути наданi гарантiї того, що дослiдження будуть завершенi, i одразу пiсля завершення дослiджень у разi невiдповiдностi специфiкацiям або наявної ймовiрностi вiдхилень вiд специфiкацiй наприкiнцi термiну придатностi отриманi данi будуть поданi до Центру (iз запропонованими заходами). |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє, включаючи опис, детальне
зображення та склад контейнера або
закупорювального матерiалу, оновленi коротку
характеристику лiкарського засобу та iнструкцiю
для медичного застосування (за необхiдностi). 2. Зразок нового пакування (за необхiдностi). 3. Дослiдження з ревалiдацiї проводилися для стерильних лiкарських засобiв, якi пройшли кiнцеву стерилiзацiю. Слiд зазначити номери серiй лiкарського засобу, що використовувались в дослiдженнях (за необхiдностi). 4. У разi змiни вiльного простору над лiкарським засобом або змiни спiввiдношення поверхня/об'єм має бути надане пiдтвердження того, що дослiдження стабiльностi розпочато вiдповiдно до вимог настанови щодо випробування стабiльностi або керiвних принципiв IСН щодо випробування стабiльностi (чинне видання) (iз зазначенням номерiв серiй лiкарського засобу) та (за необхiдностi) що на дату подання заяви про внесення змiн типу IА або IБ у розпорядженнi заявника було мiнiмум, який вимагається, даних щодо стабiльностi, а також наявнi данi не вказували на будь-яку проблему, пов'язану зi стабiльнiстю. Мають бути наданi гарантiї, що дослiдження будуть завершенi, i у разi невiдповiдностi специфiкацiям або наявної ймовiрностi вiдхилень вiд специфiкацiй наприкiнцi термiну придатностi, одразу пiсля завершення дослiджень данi будуть поданi до Центру (iз запропонованими заходами). |
| 2.2.5.5. Змiна розмiру упаковки готового лiкарського засобу | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) змiна кiлькостi одиниць (наприклад, таблеток, ампул та iн.) в упаковцi: | |||
| змiна у дiапазонi затверджених розмiрiв упаковки | 1, 2 | 1, 3 | IА |
| змiна поза дiапазоном затверджених розмiрiв упаковки | 1, 2, 3 | IБ | |
| б) вилучення упаковки певного розмiру | 3 | 1, 2 | IА |
| в) змiна маси/об'єму вмiсту контейнера багатодозового стерильного лiкарського засобу (або однодозового часткового використання) та багатодозового лiкарського засобу бiологiчного/iмунологiчного походження для парентерального застосування | II | ||
| г) змiна маси/об'єму вмiсту контейнера багатодозового лiкарського засобу для непарентеральних застосувань (або однодозового часткового використання) | 1, 2, 3 | IБ |
| Умови |
| 1. Новий розмiр упаковки повинен
вiдповiдати дозуванню i тривалостi лiкування
вiдповiдно до затвердженої короткої
характеристики лiкарського засобу. 2. Первинний пакувальний матерiал не змiнився. 3. Незмiненi форми випуску лiкарського засобу повиннi вiдповiдати iнструкцiї з дозування та тривалостi лiкування, як вказано в короткiй характеристицi лiкарського засобу. |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє, включаючи оновлену коротку
характеристику лiкарського засобу (за
необхiдностi). 2. Обґрунтування нового/незмiненого розмiру упаковки та пiдтвердження, що новий/незмiнений розмiр упаковки вiдповiдає схемi дозування та тривалостi лiкування, затвердженим у короткiй характеристицi лiкарського засобу. 3. Заява, що дослiдження стабiльностi будуть проводитись вiдповiдно до настанови щодо випробування стабiльностi або керiвних принципiв IСН щодо випробування стабiльностi (чинне видання), якщо показники стабiльностi будуть змiненi. Данi мають бути поданi до Центру лише у разi невiдповiдностi специфiкацiям (з вiдповiдною пропозицiєю). |
| Примiтка до пiдпунктiв "в" та "г" пункту 2.2.5.5. Будь-якi змiни у силi дiї лiкарського засобу потребують подання заяви про реєстрацiю лiкарського засобу. |
| 2.2.5.6. Змiна будь-якої частини матерiалу первинної упаковки, що не контактує з готовим лiкарським засобом (наприклад, колiр кришечок з контролем першого вiдкриття, колiр кодових кiлець на ампулах, контейнера для голок (рiзнi види пластмаси) | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) змiна, яка впливає на коротку характеристику лiкарського засобу | 1 | 1 | IА |
| б) змiна, яка не впливає на коротку характеристику лiкарського засобу | 1 | 1 | IА |
| Умови |
| 1. Змiна не стосується тiєї частини пакувального матерiалу, яка б могла вплинути на доставку, застосування, безпеку та стабiльнiсть готового лiкарського засобу. |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв реєстрацiйного досьє, включаючи оновлену коротку характеристику лiкарського засобу (за необхiдностi). |
| 2.2.5.7. Змiна постачальника пакувальних матерiалiв або комплектуючих (якщо зазначено в досьє) | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) вилучення постачальника | 1 | 1 | IА |
| б) замiна або додавання постачальника | 1, 2, 3, 4 | 1, 2, 3 | IА |
| в) будь-яка змiна постачальника спейсерiв для дозованих iнгаляторiв | II |
| Умови |
| 1. Жодних вилучень у компонентах
упаковки або комплектуючих не вiдбулося. 2. Кiлькiсний та якiсний склад пакувального матерiалу/комплектуючих та проектнi специфiкацiї не змiнилися. 3. Специфiкацiї та методи контролю якостi - принаймнi iдентичнi. 4. Метод та умови стерилiзацiї залишаються незмiнними (за необхiдностi). |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє. 2. Для комплектуючих для лiкарських засобiв - пiдтвердження CE-маркування (сертифiкат CE). 3. Порiвняльна таблиця затвердженої та запропонованої специфiкацiй, якщо необхiдно. |
| Примiтка. CE-маркування (CE-marking) - скорочення вiд Conformit Europeane (Європейська вiдповiднiсть) - особливий знак, який наноситься на вирiб i засвiдчує, що даний вирiб вiдповiдає основним вимогам Європейських Директив, а також те, що вiн пройшов процедуру оцiнки вiдповiдностi Директивам. Маркування "CE" вказує на те, що вирiб є безпечним для здоров'я людини, а також для навколишнього середовища. |
| 2.2.6. Стабiльнiсть |
| 2.2.6.1. Змiна у термiнах придатностi або умовах зберiгання готового лiкарського засобу: | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) зменшення термiну придатностi готового лiкарського засобу: | |||
| для торговельної упаковки | 1 | 1, 2, 3 | IА |
| пiсля першого розкриття | 1 | 1, 2, 3 | IА |
| пiсля розчинення або вiдновлення | 1 | 1, 2, 3 | IА |
| б) збiльшення термiну придатностi готового лiкарського засобу: | |||
| для торговельної упаковки (пiдтверджується даними стабiльностi реального часу) | 1, 2, 3 | IБ | |
| пiсля першого розкриття (пiдтверджується даними стабiльностi реального часу) | 1, 2, 3 | IБ | |
| пiсля розчинення або вiдновлення (пiдтверджується даними стабiльностi реального часу) | 1, 2, 3 | IБ | |
| збiльшення термiну придатностi на основi екстраполяцiї результатiв дослiдження стабiльностi, проведених не у вiдповiдностi до вимог настанови щодо дослiдження стабiльностi лiкарських засобiв або керiвних принципiв ICH щодо дослiдження стабiльностi лiкарських засобiв* | II | ||
| збiльшення термiну придатностi лiкарського засобу бiологiчного/iмунологiчного походження на основi результатiв дослiджень стабiльностi, проведених вiдповiдно до затвердженого протоколу стабiльностi | 1, 2, 3 | IБ | |
| в) змiна в умовах зберiгання лiкарського засобу бiологiчного походження, якщо дослiдження стабiльностi були проведенi не у вiдповiдностi до затвердженого протоколу стабiльностi | II | ||
| г) змiна в умовах зберiгання готового лiкарського засобу або пiсля розчинення/вiдновлення | 1, 2, 3 | IБ |
| Умови |
| 1. Змiна, не обумовлена непередбаченими обставинами у процесi виробництва або пересторогами щодо стабiльностi. |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє повиннi мiстити результати
дослiджень стабiльностi у реальному часi (що
включають повний термiн придатностi), проведених
вiдповiдно до настанови щодо дослiдження
стабiльностi лiкарських засобiв або керiвних
принципiв ICH щодо дослiдження стабiльностi
лiкарських засобiв (чинне видання) принаймнi на
двох дослiдно-промислових серiях1 готового
лiкарського засобу у затвердженiй упаковцi та/або
пiсля першого розкриття, або вiдновлення (за
необхiдностi). За потреби мають бути включенi
результати мiкробiологiчних дослiджень. 2. Оновленi: коротка характеристика лiкарського засобу, iнструкцiя для медичного застосування та пропозицiї до маркування на упаковцi. 3. Копiя затвердженої специфiкацiї наприкiнцi термiну придатностi готового лiкарського засобу та, якщо необхiдно, специфiкацiї на лiкарський засiб пiсля розчинення/вiдновлення або першого розкриття. |
| ____________ * Екстраполяцiя не застосовується до лiкарських засобiв бiологiчного/iмунологiчного походження. ____________________________________ |
| 2.2.7. Проектний простiр |
| 2.2.7.1. Введення нового проектного простору або розширення затвердженого для готового лiкарського засобу (за винятком лiкарських засобiв бiологiчного походження) щодо | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) одного або бiльше елементiв (блок, частина) виробничого процесу готового лiкарського засобу, включаючи контроль у процесi виробництва та/або методи випробувань | 1, 2, 3 | II | |
| б) методiв випробувань для допомiжних речовин/промiжних продуктiв та/або готового лiкарського засобу | 1, 2, 3 | II |
| Документацiя |
| 1. Результати дослiджень розробки
лiкарського засобу або виробничого процесу
(включаючи дослiдження оцiнки ризику та
багатомiрнi дослiдження, якщо необхiдно), якi
демонструють, що функцiональна взаємодiя
властивостей матерiалу та параметрiв процесу, якi
можуть впливати на критичнi характеристики
якостi готового лiкарського засобу, була
досягнута. 2. Опис проектного простору у формi таблицi, включаючи змiннi властивостi матерiалу та параметри процесу (якщо необхiдно) та їх запропонованi дiапазони. 3. Змiни до вiдповiдних роздiлiв матерiалiв реєстрацiйного досьє. |
| 2.2.7.2. Внесення змiн пiсля затвердження протоколу управлiння змiнами для готового лiкарського засобу | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| 1, 2 | II |
| Документацiя |
| 1. Детальний опис запропонованої змiни. 2. Протокол управлiння змiнами для готового лiкарського засобу. |
| 2.2.7.3. Вилучення затвердженого протоколу управлiння змiнами для готового лiкарського засобу | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiн |
| 1 | 1 | IА |
| Умови |
| 1. Вилучення не спричинено непередбаченими обставинами або результатами випробувань, що виходять за межi специфiкацiй пiд час внесення змiни, описаної у протоколi. |
| Документацiя |
| 1. Обґрунтування запропонованого вилучення. |
| 2.3. Сертифiкат вiдповiдностi Європейськiй фармакопеї/Сертифiкат вiдповiдностi Європейськiй фармакопеї щодо губчатої енцефалопатiї |
| 2.3.1. Подання нового або оновленого сертифiката вiдповiдностi Європейськiй фармакопеї | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| Для АФI або дiючої речовини/для вихiдного матерiалу/реагенту/промiжного продукту, що використовуються у виробництвi АФI або дiючої речовини/для допомiжної речовини | |||
| а) сертифiкат вiдповiдностi Європейськiй фармакопеї | |||
| новий сертифiкат вiдповiдностi вiд дiючого виробника | 1, 2, 3, 4, 5, 8 | 1, 2, 3, 4, 5 | IА |
| оновлений сертифiкат вiдповiдностi вiд дiючого виробника | 1, 2, 3, 4, 8 | 1, 2, 3, 4, 5 | IА |
| новий сертифiкат вiдповiдностi вiд нового виробника (замiна або доповнення) | 1, 2, 3, 4, 5, 8 | 1, 2, 3, 4, 5 | IА |
| б) сертифiкат вiдповiдностi Європейськiй фармакопеї щодо губчатої енцефалопатiї для АФI або дiючої речовини /вихiдного матерiалу/реагенту/промiжного продукту або допомiжної речовини | |||
| новий сертифiкат вiдповiдностi для АФI або дiючої речовини вiд нового або дiючого виробника | 3, 6 | 1, 2, 3, 4, 5 | IА |
| новий сертифiкат вiдповiдностi для вихiдного матерiалу/реагенту/промiжного продукту або допомiжної речовини вiд нового або дiючого виробника | 3, 6 | 1, 2, 3, 4, 5 | IА |
| оновлений сертифiкат вiдповiдностi вiд дiючого виробника | 1, 2, 3, 4, 5 | IА |
| Умови |
| 1. Специфiкацiї при випуску та наприкiнцi
термiну придатностi готового лiкарського засобу
залишаються незмiнними. 2. Додатковi (до Європейської фармакопеї) специфiкацiї щодо домiшок (за винятком залишкових розчинникiв, за умови, що вони вiдповiдають керiвним принципам IСН) та вiдповiднi вимоги до лiкарського засобу (наприклад, профiлi розмiру часток, полiморфна форма) залишаються незмiнними, за виключенням звуження (за необхiдностi). 3. Виробничий процес АФI або дiючої речовини, вихiдного матерiалу/реагенту/промiжного продукту не включає використання матерiалу людського або тваринного походження, для якого проводиться оцiнка даних щодо вiрусної безпеки. 4. Тiльки для АФI або дiючої речовини необхiдно проводити дослiдження безпосередньо перед використанням, якщо перiод повторного випробування не включений до сертифiката вiдповiдностi Європейськiй фармакопеї або якщо данi щодо перiодичностi повторного випробування ще не представленi в досьє. 5. АФI або дiюча речовина/вихiдний матерiал/реагент/промiжний продукт/допомiжна речовина - нестерильнi. 6. Для рослинних субстанцiй: шлях виробництва, фiзична форма, екстрагент та спiввiдношення лiкарський засiб/екстрагент повиннi залишатися незмiнними. |
| Документацiя |
| 1. Копiя затвердженого (оновленого)
сертифiката вiдповiдностi Європейськiй
фармакопеї. 2. У випадку додавання виробничої дiльницi у заявi слiд чiтко визначати затвердженого та запропонованого виробникiв, як зазначено у пунктi 2.5 заяви про державну реєстрацiю лiкарського засобу, форма якої наведена у додатку 1 до Порядку. 3. Змiни до вiдповiдних роздiлiв матерiалiв реєстрацiйного досьє. 4. Якщо необхiдно, документ, в якому представлена iнформацiя про будь-якi матерiали, що пiдлягають оцiнцi вiрусної безпеки вiдповiдно до керiвництва з мiнiмiзацiї ризику передачi збудникiв губчатої енцефалопатiї тварин з лiкарськими засобами, включаючи тi, що використовуються при виробництвi АФI або дiючої речовини/допомiжної речовини. Iнформацiя повинна мiстити таке: найменування виробника, види та тканини тварин, з яких було отримано матерiал, країна-постачальник тваринної сировини, її використання та попереднiй дозвiл. 5. Для АФI або дiючої речовини - заява вiд уповноваженої особи (УО) кожного з виробникiв, перелiчених у заявi на внесення змiн, якщо АФI або дiюча речовина використовується як вихiдний матерiал, та заява УО кожного з виробникiв, вiдповiдальних за випуск серiї, перелiчених у заявi. Цi заяви повиннi пiдтверджувати, що виробник(и) дiючої речовини, зазначений(i) у заявi, дiє(ють) вiдповiдно до вимог належної виробничої практики для вихiдних матерiалiв (чинне видання). Узагальнена заява може бути прийнятною за певних умов (примiтка до пiдпункту 2.2.2.1). Виробництво промiжних продуктiв також вимагає заяви УО, оскiльки будь-якi змiни до сертифiкатiв на дiючу речовину та промiжнi продукти - пов'язанi. Заява УО надається лише у тому випадку, якщо порiвняно з дiючим сертифiкатом є змiна у затвердженому перелiку виробничих дiльниць. |
| 2.3.2. Змiни, пов'язанi з необхiднiстю приведення у вiдповiднiсть до монографiї Державної фармакопеї України або Європейської фармакопеї | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) змiна у специфiкацiї нефармакопейної АФI або дiючої речовини для приведення у вiдповiднiсть до вимог Державної фармакопеї України або Європейської фармакопеї | |||
| АФI або дiюча речовина | 1, 2, 3, 4, 5 | 1, 2, 3, 4, 5 | IА |
| допомiжна речовина/вихiдний матерiал для виробництва АФI або дiючої речовини | 1, 2, 4 | 1, 2, 3, 4, 5 | IА |
| б) змiна у специфiкацiї, пов'язана зi змiнами в Державнiй фармакопеї України або Європейськiй фармакопеї | 1, 2, 4, 5 | 1, 2, 3, 4 | IА |
| в) змiна у специфiкацiї, пов'язана iз замiною вимог монографiї Державної фармакопеї України на вимоги монографiї Європейської фармакопеї | 1, 4, 5 | 1, 2, 3, 4 | IА |
| Умови |
| 1. Змiна вноситься виключно для
приведення у вiдповiднiсть до вимог Державної
фармакопеї України або Європейської фармакопеї. 2. Додатковi специфiкацiї до Державної фармакопеї України або Європейської фармакопеї залишаються незмiнними (наприклад, профiль розмiру часток, полiморфна форма або, наприклад, бiопроби, агреганти). 3. Жодних значних змiн у якiсних та кiлькiсних показниках профiлю домiшок не вiдбулося, якщо специфiкацiї не звуженi. 4. Додаткова валiдацiя нового або незмiненого фармакопейного методу не потрiбна. 5. Для рослинних субстанцiй: метод виробництва, фiзична форма, екстрагент та спiввiдношення лiкарський засiб/екстрагент повиннi залишатися незмiнними. |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє. 2. Порiвняльна таблиця вимог затвердженої та оновленої специфiкацiй. 3. Данi аналiзу двох промислових серiй АФI або дiючої речовини за всiма показниками оновленої специфiкацiї. 4. Данi, якi пiдтверджують придатнiсть монографiї для контролю АФI або дiючої речовини, наприклад, порiвняння потенцiйних домiшок iз зазначеними у примiтцi до монографiї. 5. Данi аналiзiв (у формi таблиць порiвняння) двох промислових серiй готового лiкарського засобу, що мiстять дiючу речовину, яка вiдповiдає затвердженiй та оновленiй специфiкацiям, а також порiвняльнi данi профiлю розчинення принаймнi однiєї дослiдно-промислової серiї готового лiкарського засобу. Для рослинних лiкарських засобiв можуть бути прийнятнi данi щодо розпадання. |
| Примiтка. Немає необхiдностi вносити змiни, пов'язанi зi змiнами у Державнiй фармакопеї України або Європейськiй фармакопеї, у випадку, якщо вiдповiднiсть вимогам оновленої монографiї виконується протягом шести мiсяцiв з дати її публiкацiї та зроблено посилання на поточне видання Фармакопеї у досьє на затверджений лiкарський засiб. |
| 2.4. Медичнi пристрої |
| 2.4.1. Змiна пристроїв для вимiрювання дози або введення лiкарського засобу | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) додавання або замiна пристрою, який не є невiд'ємною частиною первинної упаковки | |||
| пристрiй, який має CE-маркування | 1, 2, 3 | 1, 2, 4 | IА |
| спейсер для дозованих iнгаляторiв | II | ||
| б) вилучення пристрою | 4, 5 | 1, 5 | IА |
| в) додавання або замiна пристрою, який є невiд'ємною частиною первинної упаковки | II |
| Умови |
| 1. Запропонований пристрiй для
вимiрювання дози повинен точно видавати
необхiдну дозу препарату згiдно iз затвердженим
дозуванням. Необхiдно надати результати
вiдповiдних дослiджень. 2. Новий пристрiй повинен бути сумiсним з лiкарським засобом. 3. Змiни не повиннi призвести до суттєвих виправлень у короткiй характеристицi лiкарського засобу, iнструкцiї для медичного застосування та маркуваннi. 4. Лiкарський засiб, як i ранiше, демонструє вiдтворюванiсть дози. |
| Документацiя |
| 1. Змiни до вiдповiдних роздiлiв матерiалiв
реєстрацiйного досьє, включаючи опис, детальне
зображення та склад матерiалу пристрою, та
постачальника (за необхiдностi), оновленi: коротка
характеристика лiкарського засобу, iнструкцiя для
медичного застосування та пропозицiї до
маркування на упаковцi (за необхiдностi). 2. Пiдтвердження CE-маркування (сертифiкат CE). 3. Данi, якi пiдтверджують безпеку, точнiсть дозування та сумiснiсть матерiалу пристрою та лiкарського засобу. 4. Зразки нового пристрою (за потреби). 5. Обґрунтування виключення пристрою. |
| Примiтка до пiдпункту "в" пункту 2.4.1. Будь-яка змiна, що призводить до нової лiкарської форми, потребує подання заяви про державну реєстрацiю лiкарського засобу. |
| 2.5. Змiни до реєстрацiйного посвiдчення внаслiдок iнших регуляторних процедур |
| 2.5.1. ПМФ/ВАЗФ (мастер-файл на плазму/мастер-файл на вакцинний антиген) |
| 2.5.1.1. Включення нового, оновленого або змiненого мастер-файла на плазму у реєстрацiйне досьє на лiкарський засiб (процедура 2-го етапу для мастер-файла на плазму) | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) первинне включення нового мастер-файла на плазму, що впливає на властивостi готового лiкарського засобу | II | ||
| б) первинне включення нового мастер-файла на плазму, що не впливає на властивостi готового лiкарського засобу | 1, 2, 3, 4 | IБ | |
| в) включення оновленого/змiненого мастер-файла на плазму, якщо змiни впливають на властивостi готового лiкарського засобу | 1, 2, 3, 4 | IБ | |
| г) включення оновленого/змiненого мастер-файла на плазму, якщо змiни не впливають на властивостi готового лiкарського засобу | 1 | 1, 2, 3, 4 | IA |
| Умови | |||
| 1. Оновлений або змiнений мастер-файл на плазму (далi - ПМФ) отримав сертифiкат. | |||
| Документацiя | |||
| 1. Заява, що сертифiкат на ПМФ та звiт про
проведену оцiнку, виданi уповноваженим органом,
цiлком придатнi для реєстрацiї лiкарського засобу;
власник ПМФ надав сертифiкат, звiт про оцiнку та
мастер-файл на плазму заявнику (у випадку, якщо
заявник не є власником ПМФ), сертифiкат та звiт про
оцiнку замiнює документацiю на попереднiй ПМФ для
даного реєстрацiйного посвiдчення. 2. Сертифiкат на ПМФ та звiт про проведену оцiнку, виданi уповноваженим органом. 3. Експертний висновок дає стислий опис усiх змiн, якi внесенi до сертифiкованого ПМФ, та оцiнює їх потенцiальний вплив на готовий лiкарський засiб, включаючи оцiнки специфiчного ризику. 4. Форма заяви на внесення змiн повинна чiтко визначати "затверджений" та "запропонований" сертифiкат на ПМФ (номер сертифiката) у матерiалах реєстрацiйного досьє на готовий лiкарський засiб. Заява на внесення змiни має чiтко перелiчувати також всi iншi ПМФ, на якi посилається заявник, навiть якщо вони не є предметом заяви (за необхiдностi). |
|||
| 2.5.1.2. Включення нового, оновленого або змiненого мастер-файла на вакцинний антиген у матерiали реєстрацiйного досьє на готовий лiкарський засiб (процедура 2-го етапу для ВАЗФ) | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) первинне включення нового мастер-файла на вакцинний антиген | II | ||
| б) включення оновленого або змiненого мастер-файла на вакцинний антиген, коли змiни впливають на властивостi готового лiкарського засобу | 1, 2, 3, 4 | IБ | |
| в) включення оновленого або змiненого мастер-файла на вакцинний антиген, коли змiни не впливають на властивостi готового лiкарського засобу | 1 | 1, 2, 3, 4 | IА |
| Умови |
| 1. Оновлений або змiнений мастер-файл на вакцинний антиген отримав сертифiкат вiдповiдностi. |
| Документацiя |
| 1. Заява, що сертифiкат вiдповiдностi та
звiт про поведену оцiнку ВАЗФ, виданi
уповноваженим органом, цiлком придатнi для
реєстрацiї медичного iмунобiологiчного препарату;
власник ВАЗФ надав сертифiкат вiдповiдностi та
звiт про проведену оцiнку ВАЗФ, виданi
уповноваженим органом, заявнику (коли заявник не
є власником ВАЗФ), сертифiкат та звiт про оцiнку
замiщають документацiю попереднього ВАЗФ для
даного реєстрацiйного посвiдчення. 2. Сертифiкат вiдповiдностi та звiт про оцiнку, виданi уповноваженим органом. 3. Експертний висновок дає стислий опис усiх змiн, внесених до сертифiкованого ВАЗФ, та оцiнює їх потенцiальний вплив на готовi лiкарськi засоби, включаючи оцiнку специфiчного ризику. 4. Форма заяви на внесення змiн повинна чiтко визначати "затверджений" та "запропонований" сертифiкат вiдповiдностi на ВАЗФ (номер сертифiката) у реєстрацiйному досьє. Форма заяви на внесення змiни має чiтко перелiчувати також всi iншi ВАЗФ, на якi посилається заявник, навiть якщо вони не є предметом заяви (за необхiдностi). |
| 2.5.2. Протокол управлiння змiнами |
| 2.5.2.1. Перегляд досьє з якостi для внесення змiн на вимогу компетентного органу пiсля оцiнки протоколу управлiння змiнами | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) внесення змiни не вимагає будь-яких додаткових супровiдних даних | 1 | 1, 2, 4 | IА |
| б) внесення змiни вимагає додаткових супровiдних даних | 1, 2, 3, 4 | IВ | |
| в) внесення змiни для лiкарського засобу бiологiчного/iмунологiчного походження | 1, 2, 3, 4, 5 | IВ |
| Умови |
| 1. Запропонована змiна введена цiлком вiдповiдно до затвердженого протоколу управлiння змiнами, який вимагає негайного повiдомлення пiсля затвердження. |
| Документацiя |
| 1. Посилання на затверджений протокол
управлiння змiнами. 2. Заява про те, що змiна вiдповiдає затвердженому протоколу управлiння змiнами, результати дослiдження вiдповiдають критерiям прийнятностi, зазначеним у протоколi. Додатково заява про те, що оцiнка порiвнянностi не вимагається для лiкарських засобiв бiологiчного/iмунологiчного походження. 3. Результати дослiджень, що проводилися згiдно iз затвердженим протоколом управлiння змiнами. 4. Змiни до вiдповiдних роздiлiв матерiалiв реєстрацiйного досьє. 5. Копiя затверджених специфiкацiй на АФI або дiючу речовину або готовий лiкарський засiб. |
| III. ЗМIНИ ЩОДО БЕЗПЕКИ, ЕФЕКТИВНОСТI, ФАРМАКОНАГЛЯДУ |
| 3.1. Лiкарськi засоби |
| 3.1.1. Змiна у короткiй характеристицi лiкарського засобу, iнструкцiї для медичного застосування або маркуваннi упаковок | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiн |
| а) потребує негайного внесення, але не пiзнiше 30 днiв | 1, 2, 3 | IА | |
| б) потребує внесення вiдповiдно до iнформацiї референтного/оригiнального лiкарського засобу без надання жодних нових додаткових даних заявником | 1, 2, 3 | IБ | |
| в) потребує внесення вiдповiдно до посилань на новi додатковi данi, наданi заявником | 1, 3 | II |
| Документацiя |
| 1. До супровiдного листа заяви на
внесення змiни додати обґрунтування внесення
змiн, включаючи оновленi: коротку характеристику
лiкарського засобу, iнструкцiю для медичного
застосування та пропозицiї до маркування на
упаковцi. 2. Заява про те, що запропонованi коротка характеристика лiкарського засобу, iнструкцiя для медичного застосування та пропозицiї до маркування на упаковцi - iдентичнi такiй iнформацiї на референтний/оригiнальний лiкарський засiб. 3. Оновлена iнформацiя про лiкарський засiб. |
| 3.1.2. Змiна у короткiй характеристицi лiкарського засобу, iнструкцiї для медичного застосування, маркуваннi упаковок генеричних/комбiнованих/ бiоподiбних лiкарських засобiв пiсля оцiнки тiєї ж змiни щодо референтного/оригiнального лiкарського засобу | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiн |
| а) впровадження змiн(и), щодо яких(ої) заявник не надає жодних нових додаткових даних | 1, 2 | IБ | |
| б) впровадження змiн(и), якi (яку) необхiдно надалi обґрунтувати новими додатковими даними, якi має надати заявник (наприклад, порiвняннiсть) | II |
| Документацiя |
| 1. До супровiдного листа заяви на
внесення змiни додати рiшення МОЗ щодо змiни у
короткiй характеристицi лiкарського засобу,
iнструкцiї для медичного застосування, маркуваннi
упаковок на лiкарський засiб (за необхiдностi). 2. Оновлена iнформацiя про лiкарський засiб. |
| 3.1.3. Внесення змiн(и) на вимогу МОЗ згiдно з рекомендацiями нацiональних компетентних органiв, мiжнародних органiзацiй, регуляторних агенцiй iнших країн пiсля оцiнки негайного обмеження щодо безпеки, маркування, характерного для певної фармакологiчної та/або фармакотерапевтичної групи, регулярно оновлюваного звiту з безпеки, плану управлiння ризиками, контрольних заходiв/спецiальних зобов'язань, змiн для вiдображення короткої характеристики лiкарського засобу та iнструкцiї для медичного застосування | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiн |
| а) впровадження узгоджених змiн у формулюваннi, щодо яких заявник не надає жодних нових додаткових даних | 1, 2 | IБ | |
| б) впровадження змiн, якi вимагають подальшого обґрунтування новими додатковими даними, що надаються заявником | II |
| Документацiя |
| 1. До супровiдного листа заяви на
внесення змiни додати рiшення МОЗ щодо
необхiдностi внесення змiн та вiдповiдний звiт з
оцiнки (за необхiдностi). 2. Оновлена iнформацiя про лiкарський засiб. |
| Примiтка. Заявникам пiсля отримання нової iнформацiї, яка може призвести до змiн у реєстрацiйному посвiдченнi, слiд одразу надати таку iнформацiю та вiдповiдну заяву про внесення змiн у матерiали реєстрацiйного досьє i не чекати оцiнки цих даних компетентними органами вiдповiдно до процедури, описаної вище. |
| 3.1.4. Змiни, що пов'язанi зi значними змiнами у короткiй характеристицi лiкарського засобу, iнструкцiї для медичного застосування згiдно з новими даними з якостi, доклiнiчними, клiнiчними даними та даними фармаконагляду | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiн |
| II |
| 3.1.5. Змiна у правовому статусi лiкарського засобу | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiн |
| а) для генеричних/комбiнованих/ бiоподiбних лiкарських засобiв пiсля змiни затвердженого правового статусу референтного/оригiнального лiкарського засобу | 1, 2 | IБ | |
| б) усi iншi змiни правового статусу | II |
| Документацiя |
| 1. До супровiдного листа заяви на
внесення змiни додати обґрунтування внесення
змiн з наданням документацiї, яка пiдтверджує
змiну правового статусу лiкарського засобу. 2. Оновлена iнформацiя про лiкарський засiб. |
| 3.1.6. Змiна(и) до терапевтичних показань | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiн |
| а) додавання нового терапевтичного показання або змiни затвердженого показання | II | ||
| б) вилучення терапевтичного показання | IБ |
| Примiтка. Якщо додавання або змiна терапевтичного показання має мiсце в контекстi застосування результату процедури посилання або змiн до iнформацiї на генеричний/ комбiнований/бiоподiбний лiкарський засiб пiсля оцiнки тiєї ж змiни для референтного/оригiнального лiкарського засобу, застосовуються вiдповiдно змiни 3.1.1 та 3.1.2. |
| 3.1.7. Вилучення | Умови, якi необхiдно виконати | Документи, якi мають бути представленi | Тип змiн |
| а) лiкарська форма | 1, 2 | IБ | |
| б) сила дiї | 1, 2 | IБ |
| Документацiя |
| 1. Заява, що незмiненi форми випуску
лiкарського засобу вiдповiдають iнструкцiї з
дозування та тривалостi лiкування, як вказано в
короткiй характеристицi лiкарського засобу. 2. Переглянута iнформацiя про лiкарський засiб. |
| Примiтка. У випадках, якщо на дану лiкарську форму або силу дiї отримано реєстрацiйне посвiдчення, яке вiдрiзняється вiд реєстрацiйного посвiдчення на iншi лiкарськi форми або сили дiї, строк дiї реєстрацiйного посвiдчення на попередню лiкарську форму буде припинений. |
| 3.1.8. Введення нової системи фармаконагляду | Умови, якi необхiдно виконати | Документи, якi мають бути представленi | Тип змiн |
| а) яка не пройшла оцiнки вiдповiдним нацiональним компетентним органом/Європейською медичною агенцiєю (ЕМА) для iншого продукту того ж заявника | II | ||
| б) яка пройшла оцiнки вiдповiдним нацiональним компетентним органом/iншими регуляторними агенцiями для iншого лiкарського засобу того ж заявника* | 1 | IБ | |
| Документацiя | |||
| 1. Новий детальний опис системи фармаконагляду. | |||
| ____________ * Стосується випадкiв, коли прийнятнiсть вже оцiненої системи фармаконагляду буде оцiнюватися для нового реєстрацiйного посвiдчення. |
|||
| 3.1.9. Змiни до iснуючої системи фармаконагляду, як зазначено в описi системи фармаконагляду | Умови, якi необхiдно виконати | Документи, якi мають бути представленi | Тип змiн |
| а) змiна уповноваженої особи, вiдповiдальної за фармаконагляд | 1 | 1 | IА |
| б) змiна в контактних даних уповноваженої особи, вiдповiдальної за фармаконагляд | 1 | 2 | IА |
| в) змiна до процедури пiдтримки уповноваженої особи, вiдповiдальної за фармаконагляд | 1 | 2 | IА |
| г) змiна до бази даних з безпеки (наприклад, введення нової бази даних з безпеки, включаючи перенесення зiбраних даних з безпеки та/або аналiз та повiдомлення до нової системи) | 1, 2, 3 | 2 | IА |
| ґ) змiни в контрактних домовленостях з iншими фiзичними або юридичними особами, що залученi до виконання вимог з фармаконагляду та описанi в системi фармаконагляду, а саме: якщо складається договiр субпiдряду про подання електронних повiдомлень про iндивiдуальний випадок, пов'язаний з безпекою лiкарського засобу; про ведення головних баз даних; про вiдстеження сигналу або про пiдготовку регулярно оновлюваного звiту з безпеки | 1 | 2 | IА |
| д) виключення тем, що увiйшли у письмову процедуру(и), яка(i) описує(ють) дiяльнiсть з фармаконагляду | 1 | 2 | IА |
| е) змiна дiльницi, що проводить дiяльнiсть з фармаконагляду | 1 | 2 | IА |
| є) iншi змiни до системи фармаконагляду, якi не впливають на функцiонування системи фармаконагляду (наприклад, змiни розташування основного архiву, адмiнiстративнi змiни, оновлення скорочень, змiни щодо назви функцiй/процедур) | 1 | 2 | IА |
| Умови | |||
| 1. Система фармаконагляду залишається
незмiнною. 2. Система бази даних має бути валiдована. 3. Перенесення даних з iнших систем баз даних було валiдовано. 4. Такi самi змiни до системи фармаконагляду введенi для всiх лiкарських засобiв того самого заявника (така сама кiнцева версiя опису системи фармаконагляду). |
|||
| Документацiя | |||
| 1. Остання версiя опису системи
фармаконагляду, включаючи: а) резюме нової уповноваженої особи, вiдповiдальної за фармаконагляд; б) доказ реєстрацiї уповноваженої особи, вiдповiдальної за фармаконагляд в базi ЕМА Eudravigilance, у разi наявностi (якщо контактнi данi уповноваженої особи, вiдповiдальної за фармаконагляд, не були включенi до опису системи фармаконагляду, подання переглянутої версiї опису системи фармаконагляду не вимагається, повинна подаватися лише форма заяви/повiдомлення) та; в) нове положення про заявника та уповноважену особу, вiдповiдальну за фармаконагляд, щодо їх наявностi та засоби повiдомлення про побiчнi реакцiї, якi пiдписанi новою уповноваженою особою, вiдповiдальною за фармаконагляд, та заявником, i вiдображення будь-яких наступних змiн, наприклад, до органiзацiйної схеми. 2. Остання версiя опису системи фармаконагляду та/або остання версiя додатка стосовно лiкарського засобу, якщо необхiдно. 3. Посилання на заяву/процедуру та лiкарський засiб, до яких було внесено змiни. |
|||
| Примiтка до пiдпункту "є" пункту 3.1.9. Оцiнка поданого опису системи фармаконагляду як частини нової заяви на отримання реєстрацiйного посвiдчення/розширення/змiну може призвести до необхiдностi внесення змiн у цьому описi системи фармаконагляду на вимогу нацiонального компетентного органу. У цьому випадку такi самi змiни можуть бути внесенi до опису системи фармаконагляду в iнших реєстрацiйних посвiдченнях того самого заявника шляхом подання загальної змiни типу IА. | |||
| IV. Мастер-файл на плазму (далi - ПМФ)/мастер-файл на вакцинний антиген (далi - ВАЗФ) |
| 4.1. Змiна найменування та/або мiсцезнаходження власника ВАЗФ | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| 1 | 1 | IА |
| Умови |
| 1. Власником ВАЗФ повинна залишатися одна й та сама юридична особа або фiзична особа - пiдприємець. |
| Документацiя |
| 1. Офiцiйний документ вiдповiдного компетентного уповноваженого органу, в якому зазначено нове найменування та/або нове мiсцезнаходження власника ВАЗФ. |
| 4.2. Змiна найменування та/або мiсцезнаходження власника ПМФ | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| 1 | 1 | IА |
| Умови |
| 1. Власником ПМФ повинна залишатися одна й та сама юридична особа або фiзична особа - пiдприємець. |
| Документацiя |
| 1. Офiцiйний документ вiдповiдного компетентного уповноваженого органу, в якому зазначено нове найменування та/або нове мiсцезнаходження власника ПМФ. |
| 4.3. Змiна або передача власностi на ПМФ вiд затвердженого власника новому власнику ПМФ (а саме, iншому суб'єкту господарювання) | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| 1, 2, 3, 4, 5, 6 | IА |
| Документацiя |
| 1. Документ, який мiстить персональнi данi
(найменування та мiсцезнаходження) затвердженого
власника ПМФ, а також персональнi данi особи, якiй
передано право користування ПМФ, разом iз
запропонованою датою пiдписання передачi права
користування обома сторонами. 2. Копiя останньої сторiнки сертифiката на ПМФ (сертифiкат вiдповiдностi, виданий компетентним уповноваженим органом). 3. Обґрунтування утворення нового власника (витяг з торгового (комерцiйного) реєстру, його переклад на англiйську та українську мови), пiдписане обома сторонами. 4. Пiдтвердження передачi повного комплекту матерiалiв ПМФ з дати його первинної оцiнки правонаступнику, пiдписане обома сторонами. 5. Доручення, оформлене на контактну особу, вiдповiдальну за роботу з компетентними органами та власником ПМФ, пiдписане новим власником ПМФ. 6. Лист-пiдтвердження нового власника про виконання всiх зобов'язань, якi залишились не виконаними попереднiм власником ПМФ (якщо такi є). |
| 4.4. Змiна найменування та/або мiсцезнаходження установи (закладу) вiдбору кровi/плазми | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| 1, 2 | 1, 2, 3 | IА |
| Умови |
| 1. Така установа (заклад) повинна
залишатися тiєю самою юридичною особою. 2. Така змiна є адмiнiстративною (наприклад, об'єднання, передача повноважень); змiна найменування установи (закладу) вiдбору кровi/плазми, за умови, що установи (заклади) будуть залишатись тими самими. |
| Документацiя |
| 1. Пiдписана заява про те, що така змiна не
спричиняє змiн у системi якостi установ (закладiв)
кровi. 2. Заява про те, що перелiк установ (закладiв) кровi залишається незмiнним. 3. Оновленi вiдповiднi роздiли ПМФ та додатки до нього. |
| 4.5. Замiна або додавання нової установи (закладу) вiдбору кровi/плазми, внесеної у ПМФ | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| 1, 2, 3 | IБ |
| Документацiя |
| 1. Епiдемiологiчнi данi щодо вiрусних
маркерiв, пов'язанi з установами (закладами)
вiдбору кровi/плазми, за останнi 3 роки. Для нових
установ (закладiв) кровi або у випадку, коли такi
данi ще не доступнi, надається заява про те, що
епiдемiологiчнi данi будуть представленi пiд час
наступного щорiчного їх оновлення. 2. Пiдтвердження, що установа (заклад) кровi на даний момент працює в тих самих умовах, що зазначенi у стандартному контрактi, укладеному мiж установою (закладом) кровi та власником ПМФ. 3. Оновленi вiдповiднi роздiли ПМФ та додатки до нього. |
| 4.6. Вилучення або змiна статусу (робоча/неробоча) установи (закладу), яка займається вiдбором кровi/плазми або аналiзом зразкiв донорської кровi та пулiв плазми | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| 1, 2 | 1 | IА |
| Умови |
| 1. Пiдстави для вилучення або змiни
статусу установи (закладу) не повиннi бути
пов'язанi з питаннями належної виробничої
практики. 2. Такi установи (заклади) мають дотримуватись вимог законодавства у разi змiни статусу. |
| Документацiя |
| 1. Оновленi вiдповiднi роздiли ПМФ та додатки до нього. |
| 4.7. Додавання нової установи (закладу) забору кровi, яка не внесена у ПМФ | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| II |
| 4.8. Замiна або додавання установи (закладу), яка здiйснює аналiз зразкiв донорської кровi та/або пулiв плазми, у складi установи (закладу) кровi, внесеної у ПМФ | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| 1, 2 | IБ |
| Документацiя |
| 1. Заява про те, що аналiз здiйснюється за
тiєю самою стандартною операцiйною процедурою
та/або затвердженими методами. 2. Оновленi вiдповiднi роздiли ПМФ та додатки до нього. |
| 4.9. Додавання нової установи (закладу), яка здiйснює аналiз зразкiв донорської кровi та/або пулiв плазми, не внесеної у ПМФ | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| II |
| 4.10. Замiна або додавання нової установи (закладу) кровi, в якiй здiйснюється зберiгання плазми | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| 1, 2 | IБ |
| Документацiя |
| 1. Заява про те, що установа (заклад)
кровi, в якiй здiйснюється зберiгання плазми,
здiйснюється за тiєю самою стандартною
операцiйною процедурою, яка вже була затверджена. 2. Оновленi вiдповiднi роздiли ПМФ та додатки до нього. |
| 4.11. Вилучення установи (закладу) кровi, в якiй здiйснюється зберiгання плазми | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| 1 | 1 | IА |
| Умови |
| 1. Пiдстави для вилучення установи (закладу) не повиннi бути пов'язанi з питаннями належної виробничої практики. |
| Документацiя |
| 1. Оновленi вiдповiднi роздiли ПМФ та додатки до нього. |
| 4.12. Замiна або додавання суб'єкта господарювання, який здiйснює транспортування плазми | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| 1 | IБ |
| Документацiя |
| 1. Оновленi вiдповiднi роздiли ПМФ та додатки до нього, що мiстять перелiк усiх установ (закладiв) кровi, якi користуються послугами цього суб'єкта господарювання, резюме щодо системи на мiсцях для забезпечення належних умов транспортування (час, температура, дотримання вимог належної виробничої практики), а також пiдтвердження того, що умови транспортування будуть валiдованi. |
| 4.13. Вилучення суб'єкта господарювання, який здiйснює транспортування плазми | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| 1 | 1 | IА |
| Умови |
| 1. Пiдстави для вилучення суб'єкта господарювання не повиннi бути пов'язанi з питаннями належної виробничої практики. |
| Документацiя |
| 1. Оновленi вiдповiднi роздiли ПМФ та додатки до нього. |
| 4.14. Додавання тестового набору, який має CE-маркування, для тестування окремих зразкiв донорської кровi в якостi нового тестового набору або замiни зазначеного у ПМФ | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| 1 | 1, 2 | IА |
| Умови |
| 1. Новий тестовий набiр має CE-маркування. |
| Документацiя |
| 1. Перелiк суб'єктiв господарювання, де
проводиться тестування з використанням
тестового набору. 2. Оновленi вiдповiднi роздiли ПМФ та додатки до нього, включаючи оновлену iнформацiю щодо тестування вiдповiдно до керiвництв, що мiстять вимоги до ПМФ. |
| 4.15. Додавання тестового набору, який не має CE-маркування, для тестування окремих зразкiв донорської кровi в якостi нового тестового набору або замiни зазначеного у ПМФ | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) новий тестовий набiр, який не був затверджений у ПМФ для тестування окремих зразкiв донорської кровi в iншiй установi кровi | II | ||
| б) новий тестовий набiр, який був затверджений у ПМФ для тестування окремих зразкiв донорської кровi в iншiй установi кровi | 1, 2 | IА |
| Документацiя |
| 1. Перелiк суб'єктiв господарювання, де
проводиться тестування з використанням
тестового набору, а також перелiк органiзацiй, де
даний набiр буде використовуватися. 2. Оновленi вiдповiднi роздiли ПМФ та додатки до нього, включаючи оновлену iнформацiю щодо тестування вiдповiдно до керiвництв, що мiстять вимоги до ПМФ. |
| 4.16. Змiна набору/методу, якi використовуються для тестування пулiв плазми (антитiла або антигени, або NAT-тестування (технологiя амплiфiкацiї нуклеїнових кислот)) | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| II |
| 4.17. Введення або продовження строку iнвентаризацiйної процедури для зразкiв донорської кровi | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| 1 | 1 | IА |
| Умови |
| 1. Iнвентаризацiйна процедура проводиться за бiльш строгими вимогами. |
| Документацiя |
| 1. Оновленi вiдповiднi роздiли ПМФ, включаючи обґрунтування щодо введення або продовження строку iнвентаризацiйної процедури, установи, де застосовується процедура, а також змiни у процедурi прийняття рiшень, у тому числi новi умови. |
| 4.18. Вилучення або скорочення строку iнвентаризацiйної процедури для зразкiв донорської кровi | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| 1 | IБ |
| Документацiя |
| 1. Оновленi вiдповiднi роздiли ПМФ. |
| 4.19. Замiна або додавання контейнерiв для кровi (наприклад, пляшки, пакети) | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) новi контейнери для кровi мають CE-маркування | 1, 2 | 1 | IА |
| б) новi контейнери для кровi не мають CE-маркування | II |
| Умови |
| 1. Контейнер має CE-маркування. 2. Критерiї щодо якостi кровi залишаються незмiнними для контейнера. |
| Документацiя |
| 1. Оновленi вiдповiднi роздiли ПМФ та додатки до нього, включаючи назву контейнера, найменування виробника, специфiкацiю розчину антикоагулянта, пiдтвердження CE-маркування, а також назву установи кровi, де використовується даний контейнер. |
| 4.20. Змiни у зберiганнi/транспортуваннi | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| а) умови зберiгання та/або транспортування | 1 | 1 | IА |
| б) максимальний строк зберiгання для плазми | 1, 2 | 1 | IА |
| Умови | |||
| 1. Змiни повиннi посилити умови зберiгання
та вiдповiдати положенням монографiї
Європейської фармакопеї "Плазма людини для
фракцiонування". 2. Максимальний строк зберiгання - коротший за попереднiй. |
|||
| Документацiя | |||
| 1. Оновленi вiдповiднi роздiли ПМФ та додатки до нього, включаючи детальний опис нових умов зберiгання, данi з валiдацiї умов зберiгання/транспортування та найменування установи кровi, де впроваджується змiна (за необхiдностi). | |||
| 4.21. Впровадження тестування на вiруснi маркери за умови, що таке впровадження буде мати суттєвий вплив на оцiнку вiрусної безпеки | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| II |
| 4.22. Змiни у приготуваннi пулiв плазми (наприклад, метод приготування, розмiр пулу, зберiгання зразкiв пулiв плазми) | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| 1 | IБ | ||
| Документацiя | |||
| 1. Оновленi вiдповiднi роздiли ПМФ. | |||
| 4.23. Змiни у заходах, якi будуть вжитi, якщо ретроспективно буде встановлено, що зразки донорської кровi необхiдно виключити з процесу (ретроспективнi дослiдження) | Умови, якi мають бути виконанi | Документи, якi мають бути представленi | Тип змiни |
| II |
| Додаток 12 до Порядку проведення експертизи реєстрацiйних матерiалiв на лiкарськi засоби, що подаються на державну реєстрацiю (перереєстрацiю), а також експертизи матерiалiв про внесення змiн до реєстрацiйних матерiалiв протягом дiї реєстрацiйного посвiдчення |
ЗАЯВА
про внесення змiн до реєстрацiйних матерiалiв на
лiкарський засiб (у тому числi медичний
iмунобiологiчний препарат)
| Дата надходження заяви ___ ____________ 20__ року |
N ______________ |
| Цим я заявляю, що: р немає iнших змiн, крiм тих, що вказанi у данiй заявi (виняток: iншi змiни, що подаються паралельно та вказанi у графi "Iнша(i) заява(и)" цього додатка; р змiна(и) не спричинить(ять) негативного впливу на якiсть, ефективнiсть та безпеку лiкарського засобу; р усi умови (згiдно з додатком 11 Порядку), якi стосуються змiн(и), виконано; р необхiднi документи, що стосуються змiн(и), подано; р Заявник гарантує достовiрнiсть iнформацiї, що мiститься у матерiалах реєстрацiйного досьє, а також пiдтверджує, що всi наявнi данi стосовно якостi, безпеки та ефективностi лiкарського засобу представлено в реєстрацiйному досьє; р усi внески сплачено/буде сплачено вiдповiдно до вимог чинного законодавства. Змiни будуть введенi: р з наступного виробничого
циклу/наступного випуску
М. П. |
||||
У разi ненадання матерiалiв реєстрацiйного досьє протягом трьох мiсяцiв з дати подання заяви або листа з обґрунтуванням строку у розглядi даної заяви буде вiдмовлено. Надалi така заява може бути подана до МОЗ в установленому порядку.
| Тип змiн р Тип IА р Тип IБ р Тип II
|
|
Змiни типiв IА та IБ (нижче позначте необхiдне).
У разi змiн типу II iз заяви вилучають перелiк змiн типiв IА, IБ, наведений нижче.
У разi змiн типiв IА, IБ вилучають тi змiни типiв IА, IБ, на якi не поширюється заява.
У разi змiн, якi потребують нової реєстрацiї, вилучають тi змiни типiв IА, IБ та II, на якi не розповсюджується заява.
| I. АДМIНIСТРАТИВНI ЗМIНИ | ||||
| Назва змiни | Тип змiни | |||
| р | 1.1. Змiна найменування та/або мiсцезнаходження заявника (власника реєстрацiйного посвiдчення) | IА | IБ* | |
| р | 1.2. Змiна торговельної назви лiкарського засобу | IБ | ||
| р | 1.3. Змiна назви АФI або дiючої речовини | IА | IБ* | |
| р | 1.4. Змiна найменування та/або мiсцезнаходження виробника (включаючи за необхiдностi мiсце проведення контролю якостi) або постачальника АФI або дiючої речовини/вихiдного матерiалу/реагенту/промiжного продукту, що застосовуються у виробництвi АФI або дiючої речовини (за вiдсутностi сертифiката вiдповiдностi Європейськiй фармакопеї у затвердженому реєстрацiйному досьє) | IА | IБ* | |
| 1.5. Змiна найменування та/або мiсцезнаходження виробника готового лiкарського засобу, включаючи мiсце проведення контролю якостi | ||||
| р | а) виробнича дiльниця випуску серiй | IА | IБ* | |
| р | б) усi iншi дiльницi | IА | IБ* | |
| р | 1.6. Змiна коду АТХ | IА | IБ* | |
| р | 1.7. Вилучення виробничої дiльницi (включаючи дiльницi для АФI або дiючої речовини, промiжного продукту або готового лiкарського засобу, дiльницi для проведення пакування, виробника, вiдповiдального за випуск серiй, мiсце проведення контролю серiї) або постачальника вихiдного матерiалу, реагенту або допомiжної речовини (якщо зазначено у реєстрацiйному досьє) | IА | IБ* | |
| II. ЗМIНИ ЩОДО ЯКОСТI | ||||
| 2.1. АФI або дiюча речовина | ||||
| 2.1.1. Виробництво | ||||
| 2.1.1.1. Змiна виробника вихiдного/промiжного продукту/реагенту, що використовується у виробничому процесi АФI або дiючої речовини, або змiна виробника (включаючи, де необхiдно, мiсце проведення контролю якостi) АФI або дiючої речовини (за вiдсутностi сертифiката вiдповiдностi Європейськiй фармакопеї у матерiалах реєстрацiйного досьє) | ||||
| р | а) запропонована додаткова виробнича дiльниця є дiльницею одного виробника | IА | IБ* | |
| р | б) введення нового виробника АФI або дiючої речовини з наданням матерiалiв реєстрацiйного досьє (мастер-файла) на дiючу речовину | II | ||
| р | в) запропонований виробник використовує спосiб синтезу, що суттєво вiдрiзняється вiд попереднього, або умови виробництва, якi потенцiйно можуть змiнити важливi характеристики якостi АФI або дiючої речовини, такi як якiсний та/або кiлькiсний профiль домiшок, що потребує квалiфiкацiї, або фiзико-хiмiчнi властивостi АФI або дiючої речовини, що впливають на бiодоступнiсть | II | ||
| р | г) новий виробник вихiдного продукту, для якого вимагається попередня оцiнка вiрусної безпеки та/або ризику передачi збудникiв губчатої енцефалопатiї | II | ||
| р | ґ) змiна у АФI або дiючої речовини бiологiчного походження або вихiдному матерiалi/реагентi/промiжному продуктi, що використовується для виробництва лiкарського засобу бiологiчного походження | II | ||
| р | д) змiни у методах контролю якостi АФI або дiючої речовини або додавання дiльницi для проведення контролю серiї/випробування | IА | IБ* | |
| е) iншi змiни | IА | IБ | II | |
| 2.1.1.2. Змiни в процесi виробництва АФI або дiючої речовини | ||||
| р | а) незначна змiна у процесi виробництва АФI або дiючої речовини | IА | IБ* | |
| р | б) значна змiна у процесi виробництва АФI або дiючої речовини, що може мати iстотний вплив на якiсть, безпеку або ефективнiсть лiкарського засобу | II | ||
| р | в) змiна стосується АФI або дiючої речовини бiологiчного/iмунологiчного походження або використання хiмiчних субстанцiй у виробництвi лiкарського засобу бiологiчного/iмунологiчного походження i не вiдноситься до протоколу виробництва | II | ||
| р | г) змiна у лiкарському засобi рослинного походження, що стосується однiєї з таких характеристик: джерело походження сировини, спосiб виробництва або виготовлення | II | ||
| р | ґ) незначна змiна у закритiй частинi матерiалiв реєстрацiйного досьє (мастер-файла) на АФI або дiючу речовину | IБ | ||
| д) iншi змiни | IА | IБ | II | |
| 2.1.1.3. Змiна розмiру серiї (включаючи дiапазони) АФI або дiючої речовини або промiжного продукту | ||||
| р | а) збiльшення до 10 разiв порiвняно iз затвердженим розмiром серiї | IА | IБ* | |
| р | б) зменшення обсягу виробництва | IА | IБ* | |
| р | в) змiна розмiру серiї, що потребує доведення подiбностi АФI або дiючої речовини бiологiчного/iмунологiчного походження порiвняно iз затвердженою | II | ||
| р | г) збiльшення у понад 10 разiв порiвняно iз затвердженим розмiром серiї | IБ | ||
| р | ґ) розмiр серiї АФI або дiючої речовини бiологiчного/iмунологiчного походження збiльшився/зменшився без змiни параметрiв процесу (наприклад, дублювання лiнiї) | IБ | ||
| д) iншi змiни | IА | IБ | II | |
| 2.1.1.4. Змiни випробувань або допустимих меж, що встановленi у специфiкацiях, у процесi виробництва АФI або дiючої речовини | ||||
| р | а) звуження допустимих меж | IА | IБ* | |
| р | б) додавання нового випробування та допустимих меж | IА | IБ* | |
| р | в) вилучення несуттєвого випробування | IА | IБ* | |
| р | г) розширення затверджених допустимих меж для показникiв, якi можуть iстотно вплинути на якiсть АФI або дiючої речовини | II | ||
| р | ґ) вилучення випробування, що може мати iстотний влив на загальну якiсть АФI або дiючої речовини | II | ||
| р | д) додавання або замiна випробування за результатами дослiджень з безпеки або якостi | IБ | ||
| е) iншi змiни | IА | IБ | II | |
| 2.1.1.5. Змiни у АФI або дiючiй речовинi для сезонних, передпандемiчних або пандемiчних вакцин для профiлактики грипу | ||||
| а) замiна штаму(iв) у сезонних, передпандемiчних або пандемiчних вакцинах для профiлактики грипу | II | |||
| 2.1.2. Контроль АФI або дiючої речовини | ||||
| 2.1.2.1. Змiна у параметрах специфiкацiй та/або допустимих меж, визначених у специфiкацiях на АФI або дiючу речовину, або вихiдний/промiжний продукт/реагент, що використовуються у процесi виробництва АФI або дiючої речовини | ||||
| а) звуження допустимих меж, визначених у специфiкацiї на лiкарськi засоби, що пiдлягають випуску серiї | IА | IБ* | ||
| б) звуження допустимих меж, визначених у специфiкацiї | IА | IБ* | ||
| в) доповнення специфiкацiї новим показником якостi та вiдповiдним методом випробування | IА | IБ* | ||
| г) вилучення незначного показника якостi (наприклад, вилучення застарiлого показника) | IА | IБ* | ||
| ґ) вилучення параметра специфiкацiї, який може мати суттєвий влив на якiсть АФI або дiючої речовини та/або готового лiкарського засобу | II | |||
| д) розширення допустимих меж, визначених у специфiкацiї на АФI або дiючу речовину | II | |||
| е) розширення допустимих меж, визначених у специфiкацiях на вихiднi матерiали/промiжнi продукти, якi мають iстотний вплив на якiсть АФI або дiючої речовини та/або готового лiкарського засобу | II | |||
| є) доповнення або замiна параметра специфiкацiї за результатами дослiджень з безпеки або якостi (за винятком АФI або дiючої речовини бiологiчного або iмунологiчного походження) | IБ | |||
| ж) iншi змiни | IА | IБ | II | |
| 2.1.2.2. Змiна у методах випробування АФI або дiючої речовини або вихiдного/промiжного продукту/реагенту, що використовується у процесi виробництва АФI або дiючої речовини | ||||
| а) незначнi змiни у затверджених методах випробування | IА | IБ* | ||
| б) видалення методу випробування для АФI або дiючої речовини /реагенту/промiжного продукту, якщо альтернативний метод вже затверджений | IА | IБ* | ||
| в) iншi змiни в методах випробування (включаючи замiну або доповнення) для реагенту, що не спричиняє iстотного впливу на якiсть АФI або дiючої речовини | IА | IБ* | ||
| г) змiна у бiологiчному/iмунобiологiчному/iмунохiмiчному методi випробування або методi, у якому використовується бiологiчний реагент для АФI або дiючої речовини бiологiчного походження, наприклад, пептиднi карти, глiк-карти тощо | II | |||
| ґ) iншi змiни у методах випробування (включаючи замiну або доповнення) АФI або дiючої речовини або вихiдного/промiжного продукту | IБ | |||
| д) iншi змiни | IА | IБ | II | |
| 2.1.3. Система упаковка/укупорка | ||||
| 2.1.3.1. Змiна у безпосереднiй упаковцi АФI або дiючої речовини | ||||
| а) якiснi та/або кiлькiснi змiни складу | IА | IБ* | ||
| б) якiснi та/або кiлькiснi змiни складу для стерильних та незаморожених АФI або дiючих речовин бiологiчного/iмунологiчного походження | II | |||
| в) рiдких АФI або дiючих речовин (нестерильних) | IБ | |||
| г) iншi змiни | IА | IБ | II | |
| 2.1.3.2. Змiни у параметрах специфiкацiй та/або допустимих меж, зазначених у специфiкацiях, для первинної упаковки АФI або дiючої речовини | ||||
| а) звуження допустимих меж, зазначених у специфiкацiях | IА | IБ* | ||
| б) доповнення специфiкацiї новим показником та вiдповiдним методом випробування | IА | IБ* | ||
| в) вилучення незначного показника (наприклад, вилучення застарiлого показника) | IА | IБ* | ||
| г) доповнення або замiна показника за результатами дослiджень з безпеки або якостi | IБ | |||
| ґ) iншi змiни | IА | IБ | II | |
| 2.1.3.3. Змiна в методах випробування первинної упаковки АФI або дiючої речовини | ||||
| а) незначнi змiни у затверджених методах випробування | IА | IБ* | ||
| б) iншi змiни в методах випробування (включаючи замiну або доповнення) | IА | IБ* | ||
| в) вилучення методу випробування, якщо альтернативний метод випробування вже затверджено | IА | IБ* | ||
| 2.1.4. Стабiльнiсть | ||||
| 2.1.4.1. Змiна перiоду повторних випробувань/термiну придатностi або умов зберiгання АФI або дiючої речовини (за наявностi у матерiалах реєстрацiйного досьє сертифiката вiдповiдностi Європейськiй фармакопеї, що включає перiод повторного випробування) | ||||
| а) перiод повторного випробування/термiн придатностi | ||||
| зменшення | IА | IБ* | ||
| збiльшення перiоду повторного випробування на основi екстраполяцiї результатiв дослiдження стабiльностi, проведених не у вiдповiдностi до настанови щодо випробування стабiльностi або керiвних принципiв IСН щодо випробування стабiльностi (крiм АФI або дiючих речовин бiологiчного/iмунологiчного походження) | II | |||
| збiльшення термiну придатностi АФI або дiючої речовини бiологiчного/iмунологiчного походження на основi результатiв дослiджень, виконаних не у вiдповiдностi до затвердженого протоколу стабiльностi | II | |||
| збiльшення або введення перiоду повторного випробування/термiну придатностi на основi результатiв дослiджень у реальному часi | IБ | |||
| б) умови зберiгання | ||||
| обмеження умов зберiгання | IА | IБ* | ||
| змiна умов зберiгання АФI або дiючої речовини бiологiчного/iмунологiчного походження, якщо дослiдження стабiльностi ще не проводились вiдповiдно до затвердженого протоколу стабiльностi | II | |||
| змiна умов зберiгання АФI або дiючої речовини | IБ | |||
| в) iншi змiни | IА | IБ | II | |
| 2.1.5. Проектний простiр | ||||
| 2.1.5.1. Введення нового проектного простору або розширення затвердженого для АФI або дiючої речовини щодо | ||||
| а) одного елементу (блок, частина) виробничого процесу АФI або дiючої речовини, включаючи контроль у процесi виробництва та/або методи випробування | II | |||
| б) методiв випробувань для вихiдних матерiалiв/реагентiв/промiжних продуктiв та/або АФI або дiючої речовини | II | |||
| 2.1.5.2. Внесення змiн пiсля затвердження протоколу управлiння АФI або дiючої речовини | II | |||
| 2.1.5.3. Вилучення затвердженого протоколу управлiння змiнами для АФI або дiючої речовини | IА | IБ* | ||
| 2.2. Готовий лiкарський засiб | ||||
| 2.2.1. Опис та склад | ||||
| 2.2.1.1. Змiна або додавання штампiв, потовщень або iнших маркувань, уключаючи замiну або додавання фарб для маркування лiкарського засобу | ||||
| а) змiни штампiв, потовщень або iнших маркувань | IА | IБ* | ||
| б) змiна риски, призначеної для подiлу таблетки на рiвнi дози | IБ | |||
| в) iншi змiни | IА | IБ | II | |
| 2.2.1.2. Змiна форми або розмiрiв лiкарської форми | ||||
| а) таблетки з негайним вивiльненням, капсули, супозиторiї та песарiї | IА | IБ* | ||
| б) лiкарськi форми, стiйкi до дiї шлункового соку, з модифiкованим або пролонгованим вивiльненням та дiлимi таблетки, призначенi для подiлу на рiвнi дози | IБ | |||
| в) iншi змiни | IА | IБ | II | |
| 2.2.1.3. Змiна у складi (допомiжних речовинах) готового лiкарського засобу | ||||
| а) смаковi добавки або барвники | ||||
| додавання, вилучення або замiна | IА | IБ* | ||
| збiльшення або зменшення | IА | IБ* | ||
| б) iншi допомiжнi речовини | II | |||
| будь-яка незначна змiна кiлькiсного складу допомiжних речовин у готовому лiкарському засобi | IА | IБ* | ||
| якiснi або кiлькiснi змiни щодо однiєї або декiлькох допомiжних речовин, якi можуть значно вплинути на безпеку, якiсть або ефективнiсть готового лiкарського засобу | II | |||
| змiна у лiкарському засобi бiологiчного/iмунологiчного походження | II | |||
| будь-яка нова допомiжна речовина, що включає використання матерiалiв людського або тваринного походження, для яких вимагається оцiнка даних з вiрусної безпеки або ризику передачi збудникiв губчатої енцефалопатiї | II | |||
| змiна, яка пiдтверджується дослiдженнями з еквiвалентностi | II | |||
| замiна однiєї допомiжної речовини на iншу з тими самими функцiональними характеристиками та на тому самому рiвнi | IБ | |||
| в) iншi змiни | IА | IБ | II | |
| 2.2.1.4. Змiна маси покриття лiкарських форм для перорального застосування або змiна маси оболонки капсул | ||||
| а) твердi лiкарськi форми для перорального застосування | IА | IБ* | ||
| б) лiкарськi форми, стiйкi до дiї шлункового соку, з модифiкованим вивiльненням або пролонгованої дiї, для яких покриття є вирiшальним чинником механiзму вивiльнення | II | |||
| в) iншi змiни | IА | IБ | II | |
| 2.2.1.5. Змiна концентрацiї в окремiй дозi багатодозового лiкарського засобу для парентерального застосування, коли кiлькiсть дiючої речовини на одиницю дози (тобто сила дiї) не змiнюється | II | |||
| 2.2.1.6. Вилучення контейнера з розчинником з упаковки | IБ | |||
| 2.2.2. Виробництво | ||||
| 2.2.2.1. Замiна або введення додаткової дiльницi виробництва для частини або всього виробничого процесу готового лiкарського засобу | ||||
| а) дiльниця для вторинного пакування | IА | IБ* | ||
| б) дiльниця для первинного пакування | IА | IБ* | ||
| в) дiльниця, на якiй проводяться будь-якi виробничi стадiї, за винятком випуску серiй, проведення контролю якостi та вторинного пакування, для лiкарських засобiв бiологiчного/iмунологiчного походження | II | |||
| г) дiльниця, яка вимагає проведення первинної перевiрки виробництва або перевiрки виробництва конкретного готового лiкарського засобу | II | |||
| ґ) дiльниця, на якiй проводяться будь-якi виробничi стадiї, за винятком випуску серiй, контролю якостi, первинного та вторинного пакування, для нестерильних лiкарських засобiв | IБ | |||
| д) дiльниця, на якiй проводяться будь-якi виробничi стадiї, за винятком випуску серiї, контролю якостi та вторинного пакування для стерильних лiкарських засобiв, що виробленi з використанням асептичного методу, за винятком лiкарських засобiв бiологiчного/iмунологiчного походження | IБ | |||
| е) iншi змiни | IА | IБ | II | |
| 2.2.2.2. Змiни, що стосуються випуску серiї та контролю якостi готового лiкарського засобу | ||||
| а) замiна або додавання дiльницi, на якiй здiйснюється контроль/випробування серiї | IА | IБ* | ||
| б) замiна або додавання виробника, вiдповiдального за випуск серiї | ||||
| не включаючи контроль/випробування серiї | IА | IБ* | ||
| включаючи контроль/випробування серiї | IА | IБ* | ||
| включаючи контроль/випробування серiї для лiкарського засобу бiологiчного/iмунологiчного походження та один з методiв аналiзу, що застосовується на дiльницi, є бiологiчним/iмунологiчним/iмунохiмiчним методом | II | |||
| 2.2.2.3. Змiни у процесi виробництва готового лiкарського засобу | ||||
| а) незначна змiна у процесi виробництва твердої лiкарської форми для перорального застосування з негайним вивiльненням або розчинiв для перорального застосування | IА | IБ* | ||
| б) змiна у процесi виробництва, яка може мати iстотний влив на якiсть, безпеку та ефективнiсть лiкарського засобу | II | |||
| в) лiкарський засiб є лiкарським засобом бiологiчного/iмунологiчного походження та змiна вимагає проведення порiвняльних дослiджень | II | |||
| г) ведення нестандартного методу кiнцевої стерилiзацiї | II | |||
| ґ) введення або збiльшення припустимого надлишку дiючої речовини | II | |||
| д) незначна змiна у процесi виробництва водної суспензiї для перорального застосування | IБ | |||
| е) iншi змiни | IА | IБ | II | |
| 2.2.2.4. Змiна розмiру серiї (включаючи дiапазон розмiру серiї) готового лiкарського засобу | ||||
| а) збiльшення до 10 разiв порiвняно iз затвердженим розмiром серiї | IА | IБ* | ||
| б) зменшення до 10 разiв порiвняно iз затвердженим розмiром серiї | IА | IБ* | ||
| в) змiна вимагає оцiнки порiвнянностi (проведення порiвняльних дослiджень) лiкарського засобу бiологiчного/iмунологiчного походження | II | |||
| г) змiна стосується всiх iнших лiкарських форм сукупного (комплексного) виробничого процесу | II | |||
| ґ) збiльшення розмiру серiї бiльш нiж у 10 разiв порiвняно iз затвердженим розмiром для твердих лiкарських форм з негайним вивiльненням | IБ | |||
| д) масштаб для лiкарського засобу бiологiчного/iмунологiчного походження збiльшився/зменшився без змiни виробничого процесу (наприклад, дублювання лiнiї) | IБ | |||
| е) iншi змiни | IА | IБ | II | |
| 2.2.2.5. Змiни випробувань або допустимих меж, встановлених у специфiкацiях, пiд час виробництва готового лiкарського засобу | ||||
| а) звуження допустимих меж | IА | IБ* | ||
| б) доповнення нового методу випробування та допустимих меж | IА | IБ* | ||
| в) вилучення несуттєвого випробування | IА | IБ* | ||
| г) вилучення випробування у процесi виробництва, яке може мати iстотний вплив на загальну якiсть готового лiкарського засобу | II | |||
| ґ) розширення затверджених допустимих меж для показникiв, якi можуть мати iстотний вплив на загальну якiсть готового лiкарського засобу | II | |||
| д) доповнення або замiна випробування за результатами дослiджень з безпеки або якостi | IБ | |||
| е) iншi змiни | IА | IБ | II | |
| 2.2.3. Контроль допомiжних речовин | ||||
| 2.2.3.1. Змiна параметрiв специфiкацiй та/або допустимих меж для допомiжної речовини | ||||
| а) звуження допустимих меж | IА | IБ* | ||
| б) доповнення специфiкацiї новим показником з вiдповiдним методом випробування | IА | IБ* | ||
| в) вилучення iз специфiкацiї незначного показника (наприклад, вилучення застарiлого показника) | IА | IБ* | ||
| г) змiна знаходиться поза затвердженими допустимими межами специфiкацiй | II | |||
| ґ) вилучення iз специфiкацiї показника, який може мати iстотний вплив на загальну якiсть готового лiкарського засобу | II | |||
| д) доповнення або замiна показника специфiкацiї за результатами дослiджень з безпеки або якостi (за винятком лiкарських засобiв бiологiчного та iмунологiчного походження) | IБ | |||
| е) iншi змiни | IА | IБ | II | |
| 2.2.3.2. Змiна у методах випробування допомiжної речовини | ||||
| а) незначнi змiни у затверджених методах випробувань | IА | IБ* | ||
| б) вилучення методу випробування, якщо альтернативний метод випробування вже затверджено | IА | IБ* | ||
| в) замiна бiологiчного/iмунологiчного/iмунохiмiчного методу випробування або методу, у якому використовується бiологiчний реагент | II | |||
| г) iншi змiни у методах випробування (включаючи замiну або додавання) | IБ | |||
| 2.2.3.3. Замiна джерела одержання допомiжної речовини або реактиву, що становить ризик передачi збудникiв губчатої енцефалопатiї | ||||
| а) матерiалу, що становить ризик передачi збудникiв губчатої енцефалопатiї, на матерiал рослинного або синтетичного походження | ||||
| для допомiжних речовин або реактивiв, якi не використовуються у виробництвi активної субстанцiї бiологiчного/iмунологiчного походження або лiкарського засобу бiологiчного/iмунологiчного походження | IА | IБ* | ||
| для допомiжних речовин або реактивiв, якi використовуються у виробництвi активної субстанцiї бiологiчного/iмунологiчного походження або лiкарського засобу бiологiчного/iмунологiчного походження | IБ | |||
| б) замiна або додавання речовини, що становить ризик передачi збудникiв губчатої енцефалопатiї, або замiна речовини, що становить ризик передачi збудникiв губчатої енцефалопатiї, на iншу речовину, що становить ризик передачi збудникiв губчатої енцефалопатiї, для якої немає сертифiката вiдповiдностi Європейськiй фармакопеї щодо губчатої енцефалопатiї | II | |||
| 2.2.3.4. Змiни в методi синтезу або регенерацiї нефармакопейної допомiжної речовини (за наявностi в досьє) | ||||
| а) незначнi змiни у методi синтезу або регенерацiї | IА | IБ* | ||
| б) змiни у специфiкацiї або змiна фiзико-хiмiчних властивостей допомiжної речовини, що може мати вплив на якiсть готового лiкарського засобу | II | |||
| в) допомiжна речовина є речовиною бiологiчного/iмунологiчного походження | II | |||
| г) iншi змiни | IА | IБ | II | |
| 2.2.4. Контроль готового лiкарського засобу | ||||
| 2.2.4.1. Змiна параметрiв специфiкацiй та/або допустимих меж готового лiкарського засобу | ||||
| а) звуження допустимих меж | IА | IБ* | ||
| б) звуження допустимих меж для лiкарського засобу, що пiдлягає офiцiйному випуску серiї | IА | IБ* | ||
| в) доповнення специфiкацiї новим показником з вiдповiдним методом випробування | IА | IБ* | ||
| г) вилучення незначного показника (наприклад, вилучення застарiлого показника) | IА | IБ* | ||
| ґ) змiна знаходиться поза затвердженими допустимими межами | II | |||
| д) вилучення показника, який може мати iстотний вплив на якiсть готового лiкарського засобу | II | |||
| е) доповнення або замiна показника специфiкацiї за результатами дослiджень з безпеки або якостi (за винятком лiкарських засобiв бiологiчного/iмунологiчного походження) | IБ | |||
| є) iншi змiни | IА | IБ | II | |
| 2.2.4.2. Змiна у методах випробування готового лiкарського засобу | ||||
| а) незначна змiна у затверджених методах випробування | IА | IБ* | ||
| б) вилучення методу випробування, якщо вже затверджено альтернативний метод | IА | IБ* | ||
| в) змiна у бiологiчному/iмунологiчному/iмунохiмiчному методi випробування або методi, у якому використовується бiологiчний реагент | II | |||
| г) iншi змiни у методах випробувань (включаючи замiну або доповнення) | IБ | |||
| 2.2.4.3. Змiни, якi стосуються виробничого процесу у реальному часi або випуску за параметрами для готового лiкарського засобу | II | |||
| 2.2.5. Система упаковка/укупорка | ||||
| 2.2.5.1. Змiна у первиннiй упаковцi готового лiкарського засобу | ||||
| а) якiсний та кiлькiсний склад | ||||
| твердi лiкарськi форми | IА | IБ* | ||
| м'якi та нестерильнi рiдкi лiкарськi форми | IБ | |||
| стерильнi лiкарськi засоби та лiкарськi засоби бiологiчного/iмунологiчного походження | II | |||
| змiна стосується зниження ступеня захисту оновленої упаковки, якщо наявнi вiдповiднi змiни в умовах зберiгання та/або скорочення термiну придатностi | II | |||
| б) тип контейнера | ||||
| твердi, м'якi та нестерильнi рiдкi лiкарськi форми | IБ | |||
| стерильнi лiкарськi засоби та лiкарськi засоби бiологiчного/iмунологiчного походження | II | |||
| в) iншi змiни | IА | IБ | II | |
| 2.2.5.2. Змiна параметрiв специфiкацiй та/або допустимих меж первинної упаковки готового лiкарського засобу | ||||
| а) звуження допустимих меж, визначених у специфiкацiї | IА | IБ* | ||
| б) доповнення специфiкацiї новим показником з вiдповiдним методом випробування | IА | IБ* | ||
| в) вилучення незначного показника (наприклад, вилучення застарiлого показника) | IА | IБ* | ||
| г) додавання або замiна показника за результатами дослiджень з безпеки або якостi | IБ | |||
| ґ) iншi змiни | IА | IБ | II | |
| 2.2.5.3. Змiна у методах випробування первинної упаковки готового лiкарського засобу | ||||
| а) незначнi змiни у затверджених методах випробувань | IА | IБ* | ||
| б) iншi змiни у методах випробувань (включаючи замiну або додавання) | IА | IБ* | ||
| в) вилучення методу випробування, якщо вже затверджено альтернативний | IА | IБ* | ||
| 2.2.5.4. Змiна форми або розмiру контейнера чи закупорювального засобу (первинної упаковки) | ||||
| а) нестерильнi лiкарськi засоби | IА | IБ* | ||
| б) змiна форми або розмiру основної частини пакувального матерiалу (вторинної упаковки), що може мати значний вплив на доставку, застосування, безпеку та стабiльнiсть готового лiкарського засобу | II | |||
| в) стерильнi лiкарськi засоби | IБ | |||
| 2.2.5.5. Змiна розмiру упаковки готового лiкарського засобу | ||||
| а) змiна кiлькостi одиниць (наприклад, таблеток, ампул та iн.) в упаковцi: | ||||
| змiна у дiапазонi затверджених розмiрiв упаковки | IА | IБ* | ||
| змiна поза дiапазоном затверджених розмiрiв упаковки | IБ | |||
| б) вилучення упаковки певного розмiру | IА | IБ* | ||
| в) змiна маси/об'єму вмiсту контейнера багатодозового стерильного лiкарського засобу (або однодозового часткового використання) та багатодозового лiкарського засобу бiологiчного/iмунологiчного походження для парентерального застосування | II | |||
| г) змiна маси/об'єму вмiсту контейнера багатодозового лiкарського засобу для непарентеральних застосувань (або однодозового часткового використання) | IБ | |||
| ґ) iншi змiни | IА | IБ | II | |
| 2.2.5.6. Змiна будь-якої частини матерiалу первинної упаковки, що не контактує з готовим лiкарським засобом (наприклад, колiр кришечок з контролем першого вiдкриття, колiр кодових кiлець на ампулах, контейнера для голок (рiзнi види пластмаси) | ||||
| а) змiна, яка впливає на коротку характеристику лiкарського засобу | IА | IБ* | ||
| б) змiна, яка не впливає на коротку характеристику лiкарського засобу | IА | IБ* | ||
| 2.2.5.7. Змiна постачальника пакувальних матерiалiв або комплектуючих (якщо зазначено в досьє) | ||||
| а) вилучення постачальника | IА | IБ* | ||
| б) замiна або додавання постачальника | IА | IБ* | ||
| в) будь-яка змiна постачальника спейсерiв для дозованих iнгаляторiв | II | |||
| 2.2.6. Стабiльнiсть | ||||
| 2.2.6.1. Змiна у термiнах придатностi або умовах зберiгання готового лiкарського засобу: | ||||
| а) зменшення термiну придатностi готового лiкарського засобу: | ||||
| для торговельної упаковки | IА | IБ* | ||
| пiсля першого розкриття | IА | IБ* | ||
| пiсля розчинення або вiдновлення | IА | IБ* | ||
| б) збiльшення термiну придатностi готового лiкарського засобу: | ||||
| для торговельної упаковки (пiдтверджується даними стабiльностi реального часу) | IБ | |||
| пiсля першого розкриття (пiдтверджується даними стабiльностi реального часу) | IБ | |||
| пiсля розчинення або вiдновлення (пiдтверджується даними стабiльностi реального часу) | IБ | |||
| збiльшення термiну придатностi на основi екстраполяцiї результатiв дослiдження стабiльностi, проведених не у вiдповiдностi з вимогами настанови щодо дослiдження стабiльностi лiкарських засобiв або керiвних принципiв ICH щодо дослiдження стабiльностi лiкарських засобiв | II | |||
| збiльшення термiну придатностi лiкарського засобу бiологiчного/iмунологiчного походження на основi результатiв дослiджень стабiльностi, проведених вiдповiдно до затвердженого протоколу стабiльностi | IБ | |||
| в) змiна в умовах зберiгання лiкарського засобу бiологiчного походження, якщо дослiдження стабiльностi були проведенi не у вiдповiдностi до затвердженого протоколу стабiльностi | II | |||
| г) змiна в умовах зберiгання готового лiкарського засобу або пiсля розчинення/вiдновлення | IБ | |||
| ґ) iншi змiни | IА | IБ | II | |
| 2.2.7. Проектний простiр | ||||
| 2.2.7.1. Введення нового проектного простору або розширення затвердженого для готового лiкарського засобу (за винятком лiкарських засобiв бiологiчного походження) щодо | ||||
| а) одного або бiльше елементiв (блок, частина) виробничого процесу готового лiкарського засобу, включаючи контроль у процесi виробництва та/або методи випробувань | II | |||
| б) методiв випробувань для допомiжних речовин/промiжних продуктiв та/або готового лiкарського засобу | II | |||
| 2.2.7.2. Внесення змiн пiсля затвердження протоколу управлiння змiнами для готового лiкарського засобу | II | |||
| 2.2.7.3. Вилучення затвердженого протоколу управлiння змiнами для готового лiкарського засобу | IА | IБ* | ||
| 2.3. Сертифiкат вiдповiдностi Європейськiй фармакопеї/ Сертифiкат вiдповiдностi Європейськiй фармакопеї щодо губчатої енцефалопатiї | ||||
| 2.3.1. Подання нового або оновленого сертифiката вiдповiдностi Європейськiй фармакопеї | ||||
| Для АФI або дiючої речовини/для вихiдного матерiалу/реагенту/ промiжного продукту, що використовуються у виробництвi АФI або дiючої речовини/для допомiжної речовини | ||||
| а) сертифiкат вiдповiдностi Європейськiй фармакопеї | ||||
| новий сертифiкат вiдповiдностi вiд дiючого виробника | IА | IБ* | ||
| оновлений сертифiкат вiдповiдностi вiд дiючого виробника | IА | IБ* | ||
| новий сертифiкат вiдповiдностi вiд нового виробника (замiна або доповнення) | IА | IБ* | ||
| б) сертифiкат вiдповiдностi Європейськiй фармакопеї щодо губчатої енцефалопатiї для АФI або дiючої речовини/вихiдного матерiалу/реагенту/промiжного продукту або допомiжної речовини | ||||
| новий сертифiкат вiдповiдностi для АФI або дiючої речовини вiд нового або дiючого виробника | IА | IБ* | ||
| новий сертифiкат вiдповiдностi для вихiдного матерiалу/реагенту/промiжного продукту або допомiжної речовини вiд нового або дiючого виробника | IА | IБ* | ||
| оновлений сертифiкат вiдповiдностi вiд дiючого виробника | IА | IБ* | ||
| 2.3.2. Змiни, пов'язанi з необхiднiстю приведення у вiдповiднiсть до монографiї Державної фармакопеї України або Європейської фармакопеї | ||||
| а) змiна у специфiкацiї нефармакопейної АФI або дiючої речовини для приведення у вiдповiднiсть до вимог Державної фармакопеї України або Європейської фармакопеї | ||||
| АФI або дiюча речовина | IА | IБ* | ||
| допомiжна речовина/вихiдний матерiал для виробництва АФI або дiючої речовини | IА | IБ* | ||
| б) змiна у специфiкацiях, пов'язана зi змiнами в Державнiй фармакопеї України або Європейськiй фармакопеї | IА | IБ* | ||
| в) змiна у специфiкацiях, пов'язана iз замiною вимог монографiї Державної фармакопеї України на вимоги монографiї Європейської фармакопеї | IА | IБ* | ||
| 2.4. Медичнi пристрої | ||||
| 2.4.1. Змiна пристроїв для вимiрювання дози або введення лiкарського засобу | ||||
| а) додавання або замiна пристрою, який не є невiд'ємною частиною первинної упаковки | ||||
| пристрiй, який має CE-маркування | IА | IБ* | ||
| спейсер для дозованих iнгаляторiв | II | |||
| б) вилучення пристрою | IА | IБ* | ||
| в) додавання або замiна пристрою, який є невiд'ємною частиною первинної упаковки | II | |||
| 2.5. Змiни до реєстрацiйного посвiдчення внаслiдок iнших регуляторних процедур | ||||
| 2.5.1. ПМФ/ВАЗФ (мастер-файл на плазму/мастер-файл на вакцинний антиген) | ||||
| 2.5.1.1. Включення нового, оновленого або змiненого мастер-файла на плазму у матерiали реєстрацiйного досьє на лiкарський засiб (процедура 2-го етапу для мастер-файла на плазму) | ||||
| а) первинне включення нового мастер-файла на плазму, що впливає на властивостi готового лiкарського засобу | II | |||
| б) первинне включення нового мастер-файла на плазму, що не впливає на властивостi готового лiкарського засобу | IБ | |||
| в) включення оновленого/змiненого мастер-файла на плазму, якщо змiни впливають на властивостi готового лiкарського засобу | IБ | |||
| г) включення оновленого/змiненого мастер-файла на плазму, якщо змiни не впливають на властивостi готового лiкарського засобу | IA | IБ* | ||
| 2.5.1.2. Включення нового, оновленого або змiненого мастер-файла на вакцинний антиген у матерiали реєстрацiйного досьє на готовий лiкарський засiб (процедура 2-го етапу для ВАЗФ) | ||||
| а) первинне включення нового мастер-файла на вакцинний антиген | II | |||
| б) включення оновленого або змiненого мастер-файла на вакцинний антиген, коли змiни впливають на властивостi готового лiкарського засобу | IБ | |||
| в) включення оновленого або змiненого мастер-файла на вакцинний антиген, коли змiни не впливають на властивостi готового лiкарського засобу | IA | IБ* | ||
| 2.5.2. Протокол управлiння змiнами | ||||
| 2.5.2.1. Перегляд досьє з якостi для внесення змiн на вимогу компетентного органу пiсля оцiнки протоколу управлiння змiнами | ||||
| а) внесення змiни не вимагає будь-яких додаткових супровiдних даних | IА | IБ* | ||
| б) внесення змiни вимагає додаткових супровiдних даних | IБ | |||
| в) внесення змiни для лiкарського засобу бiологiчного/iмунологiчного походження | IБ | |||
| III. ЗМIНИ ЩОДО БЕЗПЕКИ, ЕФЕКТИВНОСТI, ФАРМАКОНАГЛЯДУ | ||||
| 3.1. Лiкарськi засоби | ||||
| 3.1.1. Змiна у короткiй характеристицi лiкарського засобу, iнструкцiї для медичного застосування або маркуваннi упаковок | ||||
| а) потребує негайного внесення, але не пiзнiше 30 днiв | IА | IБ* | ||
| б) потребує внесення вiдповiдно до iнформацiї референтного/оригiнального лiкарського засобу без надання жодних нових додаткових даних заявником | IБ | |||
| в) потребує внесення вiдповiдно до посилань на новi додатковi данi, наданi заявником | II | |||
| 3.1.2. Змiна у короткiй характеристицi лiкарського засобу, iнструкцiї для медичного застосування, маркуваннi упаковок генеричних/комбiнованих/бiоподiбних лiкарських засобiв пiсля оцiнки тiєї ж змiни щодо референтного/оригiнального лiкарського засобу | ||||
| а) впровадження змiн(и), щодо яких(ої) заявник не надає жодних нових додаткових даних | IБ | |||
| б) впровадження змiн(и), якi(у) необхiдно надалi обґрунтувати новими додатковими даними, якi має надати заявник (наприклад, порiвняннiсть) | II | |||
| 3.1.3. Внесення змiн(и) на вимогу МОЗ згiдно з рекомендацiями нацiональних компетентних органiв, мiжнародних органiзацiй, регуляторних агенцiй iнших країн пiсля оцiнки негайного обмеження щодо безпеки, маркування, характерного для певної фармакологiчної та/або фармакотерапевтичної групи, регулярно оновлюваного звiту з безпеки, плану управлiння ризиками, контрольних заходiв/спецiальних зобов'язань, змiн для вiдображення короткої характеристики лiкарського засобу та iнструкцiї для медичного застосування | ||||
| a) впровадження узгоджених змiн у формулюваннi, щодо яких заявник не надає жодних нових додаткових даних | IБ | |||
| б) впровадження змiн, якi вимагають подальшого обґрунтування новими додатковими даними, що надаються заявником | II | |||
| 3.1.4. Змiни, що пов'язанi зi значними змiнами у короткiй характеристицi лiкарського засобу, iнструкцiї для медичного застосування згiдно з новими даними з якостi, доклiнiчними, клiнiчними даними та даними фармаконагляду | II | |||
| 3.1.5. Змiна у правовому статусi лiкарського засобу | ||||
| а) для генеричних/комбiнованих/бiоподiбних лiкарських засобiв пiсля змiни затвердженого правового статусу референтного/оригiнального лiкарського засобу | IБ | |||
| б) усi iншi змiни правового статусу | II | |||
| 3.1.6. Змiна(и) до терапевтичних показань | ||||
| а) додавання нового терапевтичного показання або змiни затвердженого показання | II | |||
| б) вилучення терапевтичного показання | IБ | |||
| 3.1.7. Вилучення | ||||
| а) лiкарська форма | IБ | |||
| б) сила дiї | IБ | |||
| 3.1.8. Введення нової системи фармаконагляду | ||||
| а) яка не пройшла оцiнки вiдповiдним нацiональним компетентним органом/Європейською медичною агенцiєю (ЕМА) для iншого продукту того ж заявника | II | |||
| б) яка пройшла оцiнку вiдповiдним нацiональним компетентним органом/iншими регуляторними агенцiями для iншого лiкарського засобу того ж заявника | IБ | |||
| 3.1.9. Змiни до iснуючої системи фармаконагляду, як зазначено в описi системи фармаконагляду | ||||
| а) змiна уповноваженої особи, вiдповiдальної за фармаконагляд | IА | IБ* | ||
| б) змiна в контактних даних уповноваженої особи, вiдповiдальної за фармаконагляд | IА | IБ* | ||
| в) змiна до процедури пiдтримки уповноваженої особи, вiдповiдальної за фармаконагляд | IА | IБ* | ||
| г) змiна до бази даних з безпеки (наприклад, введення нової бази даних з безпеки, включаючи перенесення зiбраних даних з безпеки та/або аналiз та повiдомлення до нової системи) | IА | IБ* | ||
| ґ) змiни в контрактних домовленостях з iншими фiзичними або юридичними особами, що залученi до виконання вимог з фармаконагляду та описанi в системi фармаконагляду, а саме: якщо складається договiр субпiдряду про подання електронних повiдомлень про iндивiдуальний випадок, пов'язаний з безпекою лiкарського засобу; про ведення головних баз даних; про вiдстеження сигналу або про пiдготовку регулярно оновлюваного звiту з безпеки | IА | IБ* | ||
| д) виключення тем, що увiйшли у письмову процедуру(и), яка(i) описує(ють) дiяльнiсть з фармаконагляду | IА | IБ* | ||
| е) змiна дiльницi, що проводить дiяльнiсть з фармаконагляду | IА | IБ* | ||
| є) iншi змiни до системи фармаконагляду, якi не впливають на функцiонування системи фармаконагляду (наприклад, змiни розташування основного архiву, адмiнiстративнi змiни, оновлення скорочень, змiни щодо назви функцiй/процедур) | IА | IБ* | ||
| ж) iншi змiни | IА | IБ | II | |
| IV. Мастер-файл на плазму (далi - ПМФ)/мастер-файл на вакцинний антиген (далi - ВАЗФ) | ||||
| 4.1. Змiна найменування та/або мiсцезнаходження власника ВАЗФ | IА | IБ* | ||
| 4.2. Змiна найменування та/або мiсцезнаходження власника ПМФ | IА | IБ* | ||
| 4.3. Змiна або передача власностi на ПМФ вiд затвердженого власника новому власнику ПМФ (а саме, iншому суб'єкту господарювання) | IА | IБ* | ||
| 4.4. Змiна найменування та/або мiсцезнаходження установи (закладу) вiдбору кровi/плазми | IА | IБ* | ||
| 4.5. Замiна або додавання нової установи (закладу) вiдбору кровi/плазми, внесеної у ПМФ | IБ | |||
| 4.6. Вилучення або змiна статусу (робоча/неробоча) установи (закладу), яка займається вiдбором кровi/плазми або аналiзом зразкiв донорської кровi та пулiв плазми | IА | IБ* | ||
| 4.7. Додавання нової установи (закладу) забору кровi, яка не внесена у ПМФ | II | |||
| 4.8. Замiна або додавання установи (закладу), яка здiйснює аналiз зразкiв донорської кровi та/або пулiв плазми, у складi установи (закладу) кровi, внесеної у ПМФ | IБ | |||
| 4.9. Додавання нової установи (закладу), яка здiйснює аналiз зразкiв донорської кровi та/або пулiв плазми, не внесеної у ПМФ | II | |||
| 4.10. Замiна або додавання нової установи (закладу) кровi, в якiй здiйснюється зберiгання плазми | IБ | |||
| 4.11. Вилучення установи (закладу) кровi, в якiй здiйснюється зберiгання плазми | IА | IБ* | ||
| 4.12. Замiна або додавання суб'єкта господарювання, який здiйснює транспортування плазми | IБ | |||
| 4.13. Вилучення суб'єкта господарювання, який здiйснює транспортування плазми | IА | IБ* | ||
| 4.14. Додавання тестового набору, який має CE-маркування, для тестування окремих зразкiв донорської кровi в якостi нового тестового набору або замiни зазначеного у ПМФ | IА | IБ* | ||
| 4.15. Додавання тестового набору, який не має CE-маркування, для тестування окремих зразкiв донорської кровi в якостi нового тестового набору або замiни зазначеного у ПМФ | ||||
| а) новий тестовий набiр, який не був затверджений у ПМФ для тестування окремих зразкiв донорської кровi в iншiй установi кровi | II | |||
| б) новий тестовий набiр, який був затверджений у ПМФ для тестування окремих зразкiв донорської кровi в iншiй установi кровi | IА | IБ* | ||
| 4.16. Змiна набору/методу, якi використовуються для тестування пулiв плазми (антитiла або антигени, або NAT-тестування (технологiя амплiфiкацiї нуклеїнових кислот) | II | |||
| 4.17. Введення або продовження строку iнвентаризацiйної процедури для зразкiв донорської кровi | IА | IБ* | ||
| 4.18. Вилучення або скорочення строку iнвентаризацiйної процедури для зразкiв донорської кровi | IБ | |||
| 4.19. Замiна або додавання контейнерiв для кровi (наприклад, пляшки, пакети) | ||||
| а) новi контейнери для кровi мають CE-маркування | IА | IБ* | ||
| б) новi контейнери для кровi не мають CE-маркування | II | |||
| 4.20. Змiни у зберiганнi/транспортуваннi | ||||
| а) умови зберiгання та/або транспортування | IА | IБ* | ||
| б) максимальний строк зберiгання для плазми | IА | IБ* | ||
| 4.21. Впровадження тестування на вiруснi маркери за умови, що таке впровадження буде мати суттєвий вплив на оцiнку вiрусної безпеки | II | |||
| 4.22. Змiни у приготуваннi пулiв плазми (наприклад, метод приготування, розмiр пулу, зберiгання зразкiв пулiв плазми) | IБ | |||
| 4.23. Змiни у заходах, якi будуть вжитi, якщо ретроспективно буде встановлено, що зразки донорської кровi необхiдно виключити з процесу (ретроспективнi дослiдження) | II | |||
____________
* Якщо одна з умов не виконується i цю змiну
неможливо вiднести до змiн II типу.
Для документiв, що подавалися на реєстрацiю у форматi чотирьох частин (Додаток 9 до Порядку), надаються:
| Змiни у частинi I реєстрацiйного досьє | o |
|
||||||
| Змiни у частинi II реєстрацiйного досьє | o | |||||||
| Змiни у частинi III реєстрацiйного досьє | o | |||||||
| Змiни у частинi IV реєстрацiйного досьє | o |
Для документiв, що подавалися на реєстрацiю у форматi загального технiчного документа (Додаток 5 до Порядку), надаються:
| Змiни у модулi 1 реєстрацiйного досьє | o |
|
||||||||
| Змiни у модулi 2 реєстрацiйного досьє | o | |||||||||
| Змiни у модулi 3 реєстрацiйного досьє | o | |||||||||
| Змiни у модулi 4 реєстрацiйного досьє | o | |||||||||
| Змiни у модулi 5 реєстрацiйного досьє | o | |||||||||
| Iнша(i) заява(и) (дайте коротке обґрунтування запропонованим змiнам або iншим змiнам, заявленим паралельно, або поновленню заяви, або розширенню заяви) |
||||||||||
| Змiст (перерахуйте всi змiни в стислiй формi) | ||||||||||
| Передумови для внесення змiн та
обґрунтування послiдовних змiн (якщо необхiдно) (надайте коротке пояснення передумовам для пропонованих змiн, а також обґрунтування у разi послiдовних змiн) |
||||||||||
| Дiюча редакцiя* | Пропонована редакцiя* |
|
|
____________
* Укажiть точну дiючу та пропоновану редакцiю або
специфiкацiю; у разi змiн, якi стосуються
маркування первинної, вторинної (за наявностi)
упаковок та iнструкцiї для медичного
застосування лiкарського засобу, пiдкреслiть або
видiлiть змiненi слова, указанi в таблицi вище, або
подайте окремим додатком.
Додайте (за необхiдностi) пропонованi тексти iнформацiї про лiкарський засiб (коротка характеристика лiкарського засобу, iнструкцiя для медичного застосування, маркування первинної та вторинної (за наявностi) упаковок) та iншi матерiали, якi обґрунтовують внесення змiн.
| Додаток 13 до Порядку проведення експертизи реєстрацiйних матерiалiв на лiкарськi засоби, що подаються на державну реєстрацiю (перереєстрацiю), а також експертизи матерiалiв про внесення змiн до реєстрацiйних матерiалiв протягом дiї реєстрацiйного посвiдчення |
|
| ЗМIНИ ВНЕСЕНО Наказ Мiнiстерства охорони здоров'я України _____________ N _________ |
Заявник, країна:
Виробник, країна:
ЗМIНИ ДО IНСТРУКЦIЇ ДЛЯ МЕДИЧНОГО ЗАСТОСУВАННЯ ЛIКАРСЬКОГО ЗАСОБУ/ЛIКАРСЬКОГО ЗАСОБУ (МЕДИЧНОГО IМУНОБIОЛОГIЧНОГО ПРЕПАРАТУ)
НАЗВА ЛIКАРСЬКОГО ЗАСОБУ
лiкарська форма, дозування, упаковка
| Попередня редакцiя | Нова редакцiя |
| Роздiл "..." | Роздiл "..." |
| Уповноважена особа, що виступає вiд iменi заявника | Печатка, пiдпис |
| Додаток 14 до Порядку проведення експертизи реєстрацiйних матерiалiв на лiкарськi засоби, що подаються на державну реєстрацiю (перереєстрацiю), а також експертизи матерiалiв про внесення змiн до реєстрацiйних матерiалiв протягом дiї реєстрацiйного посвiдчення |
Структура короткої характеристики лiкарського засобу/лiкарського засобу (медичного iмунобiологiчного препарату)
1. Назва лiкарського засобу, дозування, лiкарська форма.
2. Якiсний i кiлькiсний склад.
3. Лiкарська форма.
4. Клiнiчна iнформацiя:
4.1. Терапевтичнi показання.
4.2. Дози та спосiб застосування.
4.3. Дiти.
4.4. Протипоказання.
4.5. Особливi застереження та запобiжнi заходи при застосуваннi.
4.6. Взаємодiя з iншими лiкарськими засобами та iншi види взаємодiй.
4.7. Застосування пiд час вагiтностi та годування груддю.
4.8. Вплив на здатнiсть керувати транспортними засобами або працювати з iншими автоматизованими системами.
4.9. Побiчнi реакцiї.
4.10. Передозування.
5. Фармакологiчнi властивостi. Фармакотерапевтична група. Код АТХ:
5.1. Фармакодинамiчнi властивостi.
5.2. Фармакокiнетичнi властивостi.
5.3. Доклiнiчнi данi з безпеки.
6. Фармацевтична iнформацiя:
6.1. Допомiжнi речовини.
6.2. Основнi випадки несумiсностi.
6.3. Термiн придатностi.
6.4. Особливi запобiжнi заходи при зберiганнi.
6.5. Тип та вмiст первинної упаковки.
6.6. Спецiальнi заходи безпеки при поводженнi з невикористаним лiкарським засобом або вiдходами лiкарського засобу (у разi необхiдностi).
7. Власник реєстрацiйного посвiдчення.
Виробник лiкарського засобу.
8. Номер реєстрацiйного посвiдчення.
9. Дата першої реєстрацiї лiкарського засобу.
10. Дата останнього перегляду.
| Додаток 15 до Порядку проведення експертизи реєстрацiйних матерiалiв на лiкарськi засоби, що подаються на державну реєстрацiю (перереєстрацiю), а також експертизи матерiалiв про внесення змiн до реєстрацiйних матерiалiв протягом дiї реєстрацiйного посвiдчення |
|
| Генеральному директору Державного експертного центру МОЗ |
|
| |
ЛИСТ
| Заявник (представник заявника) | |
, в особi | |
, |
| (найменування) | (прiзвище та iнiцiали) |
| гарантує, що при наданнi на державну реєстрацiю такого лiкарського засобу: |
| |
, |
| (назва лiкарського засобу iз зазначенням лiкарської форми) |
| на який надаються матерiали реєстрацiйного
досьє, мною додержано вимоги частини
чотирнадцятої статтi 9 Закону України "Про
лiкарськi засоби", а саме: права третьої
сторони, захищенi патентом або переданi за
лiцензiєю, не порушуються у зв'язку iз реєстрацiєю
вказаного лiкарського засобу. Менi вiдомо, що у державнiй реєстрацiї даного лiкарського засобу може бути вiдмовлено у разi, коли внаслiдок такої реєстрацiї будуть порушенi захищенi патентом чиннi майновi права iнтелектуальної власностi, у тому числi при виробництвi, використаннi, продажу лiкарських засобiв. |
| Дата | ----------------------------- | _________________________ (прiзвище та iнiцiали) |
| М. П. |
| Додаток 16 до Порядку проведення експертизи реєстрацiйних матерiалiв на лiкарськi засоби, що подаються на державну реєстрацiю (перереєстрацiю), а також експертизи матерiалiв про внесення змiн до реєстрацiйних матерiалiв протягом дiї реєстрацiйного посвiдчення |
Перелiк допомiжних речовин, якi мають бути обов'язково зазначенi на упаковцi, та iнформацiя щодо впливу цих речовин залежно вiд шляху введення та вмiсту допомiжної речовини, що має бути наведена в iнструкцiї для медичного застосування лiкарського засобу
| Назва допомiжної речовини | Код речовини | Шлях введення | Граничний вмiст | Iнформацiя в iнструкцiї для медичного застосування |
| Азобарвники: тартразин жовтий захiд FCF азорубiн, кармоїзин амарант понсо 4R (пунцовий 4R), кошенiль червона A дiамантовий чорний BN, чорний PN |
E 102 E 110 E 122 E 123 E 124 E 151 |
Пероральний | Нульовий | Може спричиняти алергiчнi реакцiї |
| Апротинiн | Зовнiшньо | Нульовий | Може спричиняти гiперчутливiсть та тяжкi алергiчнi реакцiї | |
| Аспартам | E 951 | Пероральний | Нульовий | Є похiдним фенiлаланiну, що являє небезпеку для хворих на фенiлкетонурiю |
| Бензалконiю хлорид | Для застосування в офтальмологiї | Нульовий | Може спричиняти подразнення. Необхiдно уникати контакту з м'якими контактними лiнзами (зняти контактнi лiнзи перед нанесенням препарату та встановити через 15 хв. пiсля застосування). Знебарвлює м'якi контактнi лiнзи | |
| Зовнiшньо | Спричиняє подразнення, може спричинити шкiрнi реакцiї | |||
| Iнгаляцiйний | 10 мкг/доза | Може спричинити бронхоспазм | ||
| Бергамотова олiя Бергаптен |
Зовнiшньо | Нульовий | Може пiдвищувати чутливiсть до ультрафiолетового випромiнювання (природного та штучного) | |
| Бронопол | Зовнiшньо | Нульовий | Може спричинити мiсцевi шкiрнi реакцiї (наприклад, контактний дерматит) | |
| Бутилгiдрокситолуол | E 321 | Зовнiшньо | Нульовий | Може спричинити мiсцевi шкiрнi реакцiї (наприклад, контактний дерматит) або подразнення очей та слизових оболонок |
| Бутилгiдроксiанiзол | E 320 | Зовнiшньо | Нульовий | Може спричинити мiсцевi шкiрнi реакцiї (наприклад, контактний дерматит) або подразнення очей та слизових оболонок |
| Галактоза | Парентеральний | Нульовий | Якщо у Вас встановлена непереносимiсть деяких цукрiв, проконсультуйтеся з лiкарем, перш нiж приймати цей лiкарський засiб | |
| Пероральний | Нульовий | Якщо у Вас встановлена непереносимiсть деяких цукрiв, проконсультуйтеся з лiкарем, перш нiж приймати цей лiкарський засiб | ||
| Пероральний Парентеральний |
5 г | |||
| Гепарин (як допомiжна речовина) | Парентеральний | Нульовий | Може спричинити алергiчнi реакцiї та вплинути на систему згортання кровi. Пацiєнтам з наявнiстю в анамнезi гепарин-iндукованих алергiчних реакцiй слiд уникати застосування лiкарських засобiв, що мiстять гепарин | |
| Глiцерин (глiцерол) | Пероральний | 10 г/доза | Може спричинити головний бiль, подразнення ШКТ та дiарею | |
| Ректальний | 1 г | Може чинити м'яку послаблюючу дiю | ||
| Глюкоза | Пероральний | Нульовий | Якщо у Вас встановлена непереносимiсть деяких цукрiв, проконсультуйтеся з лiкарем, перш нiж приймати цей лiкарський засiб | |
| Пероральний Парентеральний |
5 г | Мiстить ... г глюкози на дозу. З обережнiстю застосовують хворим на цукровий дiабет | ||
| Рiдкi лiкарськi форми для перорального застосування, пастилки, льодяники, жувальнi таблетки | Нульовий | Може бути шкiдливим для зубiв | ||
| Диметилсульфоксид | Зовнiшньо | Нульовий | Може спричинити подразнення шкiри | |
| Етанол (спирт) | Пероральний та парентеральний | Менше 100 мг/доза | Цей лiкарський засiб мiстить невелику кiлькiсть етанолу (алкоголю), менше 100 мг/дозу | |
| 100 мг - 3 г/доза | Цей лiкарський засiб мiстить ... об. % етанолу
(алкоголю), тобто ... мг/дозу, що еквiвалентно ... мл
пива, ... мл вина у дозi. Шкiдливий для пацiєнтiв, хворих на алкоголiзм. Слiд бути обережним при застосуваннi вагiтним та жiнкам, якi годують груддю, дiтям та пацiєнтам iз захворюваннями печiнки та хворим на епiлепсiю |
|||
| Пероральний та парентеральний | 3 г/доза | Цей лiкарський засiб мiстить ... об. % етанолу
(алкоголю), тобто... мг/дозу, що еквiвалентно ... мл
пива, ... мл вина у дозi. Шкiдливий для пацiєнтiв, хворих на алкоголiзм. Слiд бути обережним при застосуваннi вагiтним та жiнкам, якi годують груддю, дiтям та пацiєнтам iз захворюваннями печiнки та хворим на епiлепсiю. Кiлькiсть алкоголю у цьому лiкарському засобi може впливати на дiю iнших препаратiв. Кiлькiсть алкоголю у цьому лiкарському засобi може впливати на здатнiсть керувати транспортними засобами або працювати з механiзмами |
||
| Калiю сполуки | Парентеральний | Менше 1 ммоль/доза |
Цей лiкарський засiб мiстить менше 1 ммоль (39 мг)/дозу калiю, тобто практично вiльний вiд калiю | |
| Парентеральний Пероральний |
1 ммоль/доза | Цей лiкарський засiб мiстить ... ммоль (або ... мг)/дозу калiю. Слiд бути обережним при застосуваннi пацiєнтам зi зниженою функцiєю нирок або таким, хто застосовує калiй-контрольовану дiєту | ||
| Парентеральний (внутрiшньовенний) | 30 ммоль/л | Може спричинити бiль у мiсцi введення | ||
| Кислота бензойна та бензоати: кислота бензойна натрiю бензоат калiю бензоат |
E 210 E 211 E 212 |
Зовнiшньо | Нульовий | Помiрно подразнює шкiру, очi та слизовi оболонки |
| Парентеральний | Нульовий | Може пiдвищувати ризик виникнення жовтяницi у новонароджених | ||
| Кислота сорбiнова та її солi | Зовнiшньо | Нульовий | Може спричинити мiсцевi шкiрнi реакцiї (наприклад контактний дерматит) | |
| Крохмаль пшеничний | Пероральний | Нульовий | Можна застосовувати хворим на целiакiю. Пацiєнти з алергiєю на пшеницю (вiдмiнну вiд целiакiї) не повиннi застосовувати цей лiкарський засiб | |
| Ксилiт | Пероральний | 10 г | Може чинити послаблюючу дiю. Енергетична цiннiсть 1 г ксилiту - 2,4 ккал | |
| Лактит | E 966 | Пероральний | Нульовий | Якщо у Вас встановлена непереносимiсть деяких цукрiв, проконсультуйтеся з лiкарем, перш нiж приймати цей лiкарський засiб |
| 10 г | Може чинити м'яку послаблюючу дiю. Енергетична цiннiсть 1 г лактиту - 2,1 ккал | |||
| Лактоза | Пероральний | Нульовий | Якщо у Вас встановлена непереносимiсть деяких цукрiв, проконсультуйтеся з лiкарем, перш нiж приймати цей лiкарський засiб | |
| 5 г | Мiстить ... г лактози (.../2 г глюкози та .../2 г галактози) на дозу. З обережнiстю застосовують хворим на цукровий дiабет | |||
| Ланолiн | Зовнiшньо | Нульовий | Може спричинити мiсцевi шкiрнi реакцiї (наприклад, контактний дерматит) | |
| Латекс Натуральний каучук |
Будь-який | Нульовий | Упаковка цього лiкарського засобу мiстить латексну гуму. Може спричиняти тяжкi алергiчнi реакцiї | |
| Мальтит Iзомальтит Мальтит рiдкий (сироп глюкози гiдрогенiзований) |
E 965 E 953 |
Пероральний | Нульовий | Якщо у Вас встановлена непереносимiсть деяких цукрiв, проконсультуйтеся з лiкарем, перш нiж приймати цей лiкарський засiб |
| 10 г | Може чинити м'яку послаблюючу дiю. Енергетична цiннiсть 1 г мальтиту (або iзомальтиту) - 2,3 ккал | |||
| Манiт | E 421 | Пероральний | 10 г | Може чинити м'яку послаблюючу дiю |
| Натрiю сполуки | Парентеральний | Менше 1 ммоль/доза | Цей лiкарський засiб мiстить менше 1 ммоль (23 мг)/дозу натрiю, тобто практично вiльний вiд натрiю | |
| Пероральний Парентеральний |
1 ммоль/доза | Цей лiкарський засiб мiстить ... ммоль (або ... мг)/дозу натрiю. Слiд бути обережним при застосуваннi пацiєнтам, якi застосовують натрiй-контрольовану дiєту | ||
| Олiя арахiсова | Будь-який | Нульовий | Лiкарський засiб мiстить арахiсову олiю. Якщо у Вас є алергiя на арахiс або сою, не вживайте цей лiкарський засiб | |
| Олiя кунжутна | Будь-який | Нульовий | Рiдко може спричиняти тяжкi алергiчнi реакцiї | |
| Олiя рицинова полiетоксильована та олiя рицинова полiетоксильована, гiдрогенiзована | Парентеральний | Нульовий | Може спричинити тяжкi алергiчнi реакцiї | |
| Пероральний | Нульовий | Може спричинити розлад шлунка та дiарею | ||
| Зовнiшньо | Нульовий | Може спричинити шкiрнi реакцiї | ||
| Олiя соєва та олiя соєва гiдрогенiзована | Будь-який | Нульовий | Лiкарський засiб мiстить соєву олiю. Якщо у Вас є алергiя на арахiс або сою, не вживайте цей лiкарський засiб | |
| Органiчнi сполуки ртутi, наприклад: тiомерсал фенiлртутi нiтрат фенiлртутi ацетат фенiлртутi борат |
Для застосування в офтальмологiї | Нульовий | Може спричинити алергiчнi реакцiї | |
| Зовнiшньо | Нульовий | Може спричинити мiсцевi шкiрнi реакцiї (наприклад контактний дерматит) та знебарвлення шкiри | ||
| Парентеральний | Нульовий | Цей лiкарський засiб мiстить тiомерсал як консервант та може спричинити у Вас/Вашої дитини алергiчну реакцiю. Повiдомте лiкаря, якщо у Вас/Вашої дитини були/виникли алергiчнi реакцiї | ||
| Парагiдроксибензоати та їх ефiри: етилпарагiдроксибензоат пропiлпарагiдроксибензоат натрiю пропiлпарагiдрокси- бензоат метилпарагiдроксибензоат натрiю метилпарагiдроксибензоат |
E 214 E 216 E 217 E 218 E 219 |
Пероральний Для застосування в офтальмологiї Зовнiшньо |
Нульовий | Може спричинити алергiчнi реакцiї (можливо, уповiльненi) |
| Парентеральний Iнгаляцiйний |
Нульовий | Може спричинити алергiчнi реакцiї (можливо, уповiльненi) та в окремих випадках бронхоспазм | ||
| Перуанський бальзам | Зовнiшньо | Нульовий | Може спричинити шкiрнi реакцiї | |
| Пропiленглiколь та ефiри | Зовнiшньо | Нульовий | Може спричинити подразнення шкiри | |
| Пероральний Парентеральний |
400 мг/кг (для дорослих) 200 мг/кг (для дiтей) |
Може спричинити симптоми, схожi з такими, що виникають при вживаннi алкоголю | ||
| Сорбiт | E 420 | Пероральний Парентеральний |
Нульовий | Якщо у Вас встановлена непереносимiсть деяких цукрiв, проконсультуйтеся з лiкарем, перш нiж приймати цей лiкарський засiб |
| Пероральний | 10 г | Може чинити м'яку послаблюючу дiю. Енергетична цiннiсть 1 г сорбiту - 2,6 ккал | ||
| Спирт бензиловий | Парентеральний | Менше 90 мг/кг/добу |
Не застосовують недоношеним дiтям та
новонародженим. Може спричинити токсичнi та алергiчнi реакцiї у немовлят та дiтей вiком до 3 рокiв |
|
| 90 мг/кг/добу | Не застосовують недоношеним дiтям та
новонародженим. Через ризик фатальних токсичних реакцiй не застосовувати немовлятам та дiтям вiком до 3 рокiв |
|||
| Спирт стеариловий | Зовнiшньо | Нульовий | Може спричинити мiсцевi шкiрнi реакцiї (наприклад, контактний дерматит) | |
| Спирт цетиловий | Зовнiшньо | Нульовий | Може спричинити мiсцевi шкiрнi реакцiї (наприклад, контактний дерматит) | |
| Спирт цетостеариловий | Зовнiшньо | Нульовий | Може спричинити мiсцевi шкiрнi реакцiї (наприклад, контактний дерматит) | |
| Сульфiти, включаючи метабiсульфiти, наприклад: сiрки дiоксид натрiю сульфiт натрiю гiдросульфiт натрiю метабiсульфiт калiю метабiсульфiт калiю гiдросульфiт |
E 220 E 221 E 222 E 223 E 224 E 228 |
Пероральний Парентеральний Респiраторний |
Нульовий | Рiдко може спричиняти реакцiї гiперчутливостi та бронхоспазм |
| Фенiлаланiн | Будь-який | Нульовий | Цей лiкарський засiб мiстить фенiлаланiн. Може бути небезпечним для хворих на фенiлкетонурiю |
|
| Формальдегiд | Зовнiшньо | Нульовий | Може спричинити мiсцевi шкiрнi реакцiї (наприклад контактний дерматит) | |
| Пероральний | Нульовий | Може спричинити розлад шлунка та дiарею | ||
| Фруктоза | Пероральний Парентеральний |
Нульовий | Якщо у Вас встановлена непереносимiсть деяких цукрiв, проконсультуйтеся з лiкарем, перш нiж приймати цей лiкарський засiб | |
| 5 г | Мiстить ... г фруктози на дозу. З обережнiстю застосовують хворим на цукровий дiабет | |||
| Рiдкi лiкарськi форми для перорального застосування, пастилки, льодяники, жувальнi таблетки | Нульовий | Може бути шкiдливим для зубiв | ||
| Хлоркрезол | Зовнiшньо Парентеральний |
Нульовий | Може спричиняти алергiчнi реакцiї | |
| Цукор iнвертний | Пероральний | Нульовий | Якщо у Вас встановлена непереносимiсть деяких цукрiв, проконсультуйтеся з лiкарем, перш нiж приймати цей лiкарський засiб | |
| 5 г | Мiстить ... г сумiшi фруктози та глюкози на дозу. З обережнiстю застосовують хворим на цукровий дiабет | |||
| Рiдкi лiкарськi форми для перорального застосування, пастилки, льодяники, жувальнi таблетки | Нульовий | Може бути шкiдливим для зубiв | ||
| Цукроза (сахароза) | Пероральний | Нульовий | Якщо у Вас встановлена непереносимiсть деяких цукрiв, проконсультуйтеся з лiкарем, перш нiж приймати цей лiкарський засiб | |
| 5 г | Мiстить ... г цукрози на дозу. З обережнiстю застосовують хворим на цукровий дiабет | |||
| Рiдкi лiкарськi форми для перорального застосування, пастилки, льодяники, жувальнi таблетки | Нульовий | Може бути шкiдливим для зубiв |
____________
Термiн "нульовий" означає будь-який вмiст
речовини, починаючи з мiнiмального.
| ЗАТВЕРДЖЕНО Наказ Мiнiстерства охорони здоров'я України 04.01.2013 N 3 |
|
| Зареєстровано в Мiнiстерствi юстицiї України 15 березня 2013 р. за N 426/22958 |
Положення про Квалiфiкацiйну комiсiю Мiнiстерства охорони здоров'я України
I. Загальнi положення
1.1. Квалiфiкацiйна комiсiя Мiнiстерства охорони здоров'я України (далi - Комiсiя) - консультативно-дорадчий орган, що дiє при Мiнiстерствi охорони здоров'я України (далi - МОЗ).
1.2. Комiсiя у своїй дiяльностi керується Законом України "Про лiкарськi засоби", Порядком державної реєстрацiї (перереєстрацiї) лiкарських засобiв, затвердженим постановою Кабiнету Мiнiстрiв України вiд 26 травня 2005 року N 376, iншими нормативно-правовими актами у сферi реєстрацiї лiкарських засобiв, у тому числi цим Положенням.
II. Права та обов'язки Комiсiї
2.1. Основними обов'язками Комiсiї є:
2.1.1. Розгляд висновкiв Центру щодо первинної експертизи заяви про державну реєстрацiю лiкарського засобу з точки зору його належностi до заборонених до застосування в Українi лiкарських засобiв та квалiфiкацiї типу такої заяви згiдно з вимогами Порядку проведення експертизи реєстрацiйних матерiалiв на лiкарськi засоби, що подаються на державну реєстрацiю (перереєстрацiю), а також експертизи матерiалiв про внесення змiн до реєстрацiйних матерiалiв протягом дiї реєстрацiйного посвiдчення, затвердженого наказом Мiнiстерства охорони здоров'я України вiд 26 серпня 2005 року N 426 (у редакцiї наказу Мiнiстерства охорони здоров'я України вiд 04 сiчня 2013 року N 3), зареєстрованим у Мiнiстерствi юстицiї України 19 вересня 2005 року за N 1069/11349, та надання МОЗ рекомендацiй щодо типу заяви лiкарського засобу, поданого з метою державної реєстрацiї.
2.1.2. Надання рекомендацiй МОЗ щодо визначення типу заяви поданого для державної реєстрацiї в Українi лiкарського засобу.
2.1.3. Надання рекомендацiй МОЗ щодо вiдмови в державнiй реєстрацiї (перереєстрацiї), внесеннi змiн до матерiалiв реєстрацiйного досьє лiкарського засобу за результатами попередньої експертизи матерiалiв реєстрацiйного досьє, проведеної Центром.
2.1.4. Надання рекомендацiй МОЗ щодо вiдмови в державнiй реєстрацiї (перереєстрацiї), внесеннi змiн до матерiалiв реєстрацiйного досьє лiкарського засобу за результатами спецiалiзованої експертизи матерiалiв реєстрацiйного досьє, проведеної Центром.
2.1.5. Надання рекомендацiй МОЗ щодо направлення лiкарського засобу на додатковi випробування згiдно з роздiлами XIX та XX Порядку проведення експертизи реєстрацiйних матерiалiв на лiкарськi засоби, що подаються на державну реєстрацiю (перереєстрацiю), а також експертизи матерiалiв про внесення змiн до реєстрацiйних матерiалiв протягом дiї реєстрацiйного посвiдчення, затвердженого наказом Мiнiстерства охорони здоров'я України вiд 26 серпня 2005 року N 426 (у редакцiї наказу Мiнiстерства охорони здоров'я України вiд 04 сiчня 2013 року N 3), зареєстрованим у Мiнiстерствi юстицiї України 19 вересня 2005 року за N 1069/11349, на пiдставi рекомендацiй Центру.
2.1.6. Дотримання принципiв неупередженостi, конфiденцiйностi та компетентностi.
2.2. Комiсiя розглядає висновки Центру щодо первинної експертизи заяви про державну реєстрацiю лiкарського засобу, щодо вiдмови у державнiй реєстрацiї (перереєстрацiї), внесеннi змiн до матерiалiв реєстрацiйного досьє лiкарського засобу за результатами експертизи.
2.3. Комiсiя зберiгає документи, що розглядалися нею, з питань реєстрацiї (перереєстрацiї), внесення змiн до матерiалiв реєстрацiйного досьє лiкарського засобу та вiдмов протягом п'яти рокiв пiсля оформлення протоколу Комiсiї, а потiм передає до архiву МОЗ.
2.4. Комiсiя має право:
2.4.1. Запитувати у Центру додатковi матерiали щодо реєстрацiї (перереєстрацiї), внесення змiн до матерiалiв реєстрацiйного досьє лiкарського засобу, iнформацiю про всi доповнення та змiни, що вносяться до матерiалiв реєстрацiйного досьє, вiдхилення та ускладнення, конфлiктнi ситуацiї, пов'язанi з питаннями ефективностi, безпеки та якостi лiкарського засобу (за потреби), необхiднi для виконання своїх основних обов'язкiв.
2.4.2. Подавати письмовi пропозицiї до Центру (за потреби).
2.4.3. Брати участь у роботi конференцiй, симпозiумiв, семiнарiв, шкiл стосовно аспектiв проведення реєстрацiї (перереєстрацiї), внесення змiн до матерiалiв реєстрацiйного досьє лiкарського засобу.
2.4.4. Розробляти та подавати до МОЗ пропозицiї щодо удосконалення дiяльностi Комiсiї.
III. Склад та порядок роботи Комiсiї
3.1. Персональний склад Комiсiї формує та затверджує МОЗ.
3.2. Комiсiю очолює Голова.
Голова, його заступник та вiдповiдальний секретар обираються на першому засiданнi вiдкритим голосуванням простою бiльшiстю голосiв членiв Комiсiї, присутнiх на засiданнi.
3.3. Формою роботи Комiсiї є засiдання.
Засiдання проводяться по мiрi надходження матерiалiв з Центру у строки, визначенi Порядком проведення експертизи реєстрацiйних матерiалiв на лiкарськi засоби, що подаються на державну реєстрацiю (перереєстрацiю), а також експертизи матерiалiв про внесення змiн до реєстрацiйних матерiалiв протягом дiї реєстрацiйного посвiдчення, затвердженим наказом Мiнiстерства охорони здоров'я України вiд 26 серпня 2005 року N 426 (у редакцiї наказу Мiнiстерства охорони здоров'я України вiд 04 сiчня 2013 року N 3), зареєстрованим у Мiнiстерствi юстицiї України 19 вересня 2005 року за N 1069/11349.
3.4. Засiдання є правомочним, якщо на ньому присутнi бiльше половини членiв Комiсiї.
3.5. Рiшення Комiсiї приймається вiдкритим голосуванням простою бiльшiстю голосiв членiв Комiсiї, присутнiх на засiданнi, оформляється протоколом, який пiдписують Голова та вiдповiдальний секретар. При рiвнiй кiлькостi голосiв голос Голови є вирiшальним.
На пiдставi рекомендацiй Комiсiї щодо вiдмови в державнiй реєстрацiї (перереєстрацiї), внесеннi змiн до матерiалiв реєстрацiйного досьє лiкарського засобу за результатами попередньої та спецiалiзованої експертиз, проведених Центром, МОЗ приймає рiшення, яке оформляється вiдповiдним наказом.
3.6. Органiзацiйно-технiчне забезпечення дiяльностi Комiсiї здiйснюється МОЗ.
| Начальник Управлiння лiкарських засобiв та медичної продукцiї МОЗ України | Л. В. Коношевич |
 | Copyright © 2026 НТФ «Интес» Все права сохранены. |